As filed with the Securities and Exchange Commission on January 7, 2022.
Registration No. 333-260565
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_______________________________________________
Amendment No. 5
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
_______________________________________________
Intensity Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
_______________________________________________
|
Delaware |
2836 |
46-1488089 |
||
|
(State or other jurisdiction of |
(Primary standard industrial |
(I.R.S. employer |
_______________________________________________
61 Wilton Road, 3rd Floor
Westport, CT 06880
Telephone: (203) 221-7381
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
_______________________________________________
Lewis H. Bender
Chief Executive Officer
61 Wilton Road, 3rd Floor
Westport, CT 06880
Telephone: (203) 221-7381
(Name, address, including zip code, and telephone number, including area code, of agent for service)
_______________________________________________
Copies to:
|
Robert H. Cohen, Esq. |
Ivan K. Blumenthal, Esq. |
_______________________________________________
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ☐
If this form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one).
|
Large accelerated filer |
☐ |
Accelerated filer |
☐ |
|||||
|
Non-accelerated filer |
R |
Smaller reporting company |
R |
|||||
|
Emerging growth company |
R |
If an emerging growth company, indicate by checkmark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided to Section 7(a)(2)(B) of the Securities Act. ☐
CALCULATION OF REGISTRATION FEE
|
Title of Each Class of Securities to be Registered |
Proposed |
Amount of |
||||
|
Common Stock, $0.0001 par value per share |
$ |
17,250,002 |
$ |
1,599.08 |
||
____________
(1) Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(o) of the Securities Act of 1933, as amended (the “Securities Act”).
(2) Includes the aggregate offering price of additional shares that the underwriters have the option to purchase to cover over-allotments, if any.
(3) Calculated pursuant to Rule 457(o) of the Securities Act.
(4) $1,599.08 previously paid.
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
|
Preliminary Prospectus |
Subject to Completion |
DATED January 7, 2022 |
|
2,142,858 Shares of Common Stock
|
||

This is the initial public offering of shares of common stock of Intensity Therapeutics, Inc. We are offering 2,142,858 shares of common stock. Prior to this offering, there has been no public market for our common stock. The initial public offering price per share of our common stock is expected to be between $6.00 and $8.00. We have applied to list our common stock on the Nasdaq Capital Market under the symbol “INTS.”
Unless otherwise indicated or the context otherwise requires, references in this prospectus to the “Company”, “we”, “us” and “our” refer to Intensity Therapeutics, Inc.
We are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012 and, as such, may elect to comply with certain reduced public company reporting requirements. See the section entitled “Prospectus Summary — Implications of Being an Emerging Growth Company” in this prospectus.
Investing in our common stock involves a high degree of risk. Before buying any shares, you should carefully read the discussion of the material risks of investing in our common stock under the heading “Risk Factors” beginning on page 8 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
|
Per share |
Total |
|||||
|
Initial public offering price |
$ |
|
$ |
|
||
|
Underwriting discounts and commissions(1) |
$ |
|
$ |
|
||
|
Proceeds, before expenses, to us |
$ |
|
$ |
|
||
____________
(1) See “Underwriters” beginning on page 122 of this prospectus for additional information regarding the compensation payable to the underwriters.
The underwriters have an option to purchase up to 321,428 additional shares from us at the initial public offering price, less the underwriting discounts and commissions. The underwriters can exercise this option at any time and from time to time within 45 days from the date of this prospectus.
Delivery of the shares of our common stock will be made on or about , 2022.
Sole Book-Running Manager
A.G.P.
Co-Manager
Brookline Capital Markets
a division of Arcadia Securities, LLC
The date of this Prospectus is , 2022.
|
Page |
||
|
1 |
||
|
8 |
||
|
35 |
||
|
36 |
||
|
37 |
||
|
38 |
||
|
40 |
||
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
42 |
|
|
51 |
||
|
96 |
||
|
101 |
||
|
108 |
||
|
110 |
||
|
112 |
||
|
115 |
||
|
Material U.S. Federal Income Tax Considerations for Non-U.S. Holders of Common Stock |
117 |
|
|
122 |
||
|
127 |
||
|
127 |
||
|
127 |
||
|
F-1 |
You should rely only on the information contained in this prospectus or in any free writing prospectus we may authorize to be delivered or made available to you. We have not, and the underwriters have not, authorized anyone to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are offering to sell, and seeking offers to buy, shares of our common stock only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of shares of our common stock.
For investors outside the United States: We have not, and the underwriters have not, done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the shares of common stock and the distribution of this prospectus outside the United States.
Market and Other Industry Data
Unless otherwise indicated, market data and certain industry forecasts used throughout this prospectus were obtained from various sources, including internal surveys, market research, consultant surveys, publicly available information and industry publications and surveys. Industry surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but that the accuracy and completeness of such information is not guaranteed. Such data and industry forecasts involve a number of assumptions and limitations and they are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the sections entitled “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” and elsewhere in this prospectus. These and other factors could cause results to differ materially from those expressed in these publications and reports.
i
Trademarks and Other Intellectual Property Rights
We own or have rights to trademarks or trade names that we use in connection with the operation of our business, including our corporate names, tag-lines, logos and website names. In addition, we own or have the rights to patents, copyrights, trade secrets and other proprietary rights that protect our service offerings. Solely for convenience, some of the copyrights, trade names and trademarks referred to in this prospectus are listed without their ©, ® and ™ symbols, but we will assert, to the fullest extent under applicable law, our rights to our copyrights, trade names and trademarks.
ii
The following summary highlights information appearing elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should read this entire prospectus carefully, and in particular, the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the notes relating to those statements included elsewhere in this prospectus. Some of the statements in this prospectus constitute forward-looking statements. See the section entitled “Cautionary Note Regarding Forward-Looking Statements”.
Our Company
Intensity Therapeutics, Inc. is a clinical stage biotechnology company passionately committed to applying scientific leadership in the field of localized cancer reduction leading to anti-cancer immune activation. Our new approach involves the direct injection into tumors of a unique product created from our DfuseRx℠ discovery platform.
One challenge that we have identified with current intratumoral (IT) treatment approaches is that a tumor’s lipophilic, high fat and pressurized microenvironment does not effectively absorb water-based products. We believe that this drug delivery challenge limits the effectiveness of prior and current IT treatments that formulate their product candidates by injecting aqueous products (regardless of the mechanism or approach, i.e. the stimulation of an inflammatory response or efforts to attract immune cells into a hostile live tumor). Accordingly, there remains a continued unmet need for the development of direct IT therapies for solid tumors that provide high local killing efficacy coupled with nontoxic systemic anti-cancer effects. We believe we have created a product candidate with the necessary chemistry to overcome this local delivery challenge. Evidence shows the mechanism of tumor killing achieved by our drug candidate also leads to systemic immune activation in certain cancers.
Our platform creates patented anti-cancer product candidates comprising active anti-cancer agents and amphiphilic molecules. Amphiphilic molecules have two distinct components: one part is soluble in water and the other is soluble in fat or oils. When an amphiphilic compound is mixed with therapeutic agents, such as chemotherapies, the agents also become soluble in both fat and water. Our product candidates include novel formulations consisting of potent anti-cancer drugs mixed together with these amphiphilic agents.
Our lead product candidate, INT230-6, consists of two proven anti-cancer cytotoxic agents, cisplatin and vinblastine sulfate, mixed with the amphiphilic molecule (SHAO) — all in one vial. The anti-cancer agents, cisplatin and vinblastine sulfate, used in our product candidate are both generic. These agents are available to purchase in bulk supply commercially. The United States Food & Drug Administration (the “FDA”) has approved both drugs as intravenous agents for several types of cancers. Cisplatin was first approved in 1978 for testicular cancer. Per the product labeling, cisplatin’s approved indications include treatment of testicular, ovarian and bladder cancer. The drug is also used widely in several other cancers including pancreatic and bile duct cancer. Vinblastine sulfate was first approved in 1965. Per the product labeling, vinblastine sulfate’s approved indications include treatment of generalized Hodgkin’s disease, lymphocytic lymphoma, advanced carcinoma of the testis, and Kaposi’s sarcoma. The drug is also used in breast and lung cancer. In 2017, we initiated a Phase 1/2 dose escalation study using INT230-6 in the United States under an investigational new drug application (“IND”) authorized by the FDA and in Canada following receipt of a no objection letter from Health Canada. The study, IT-01, is exploring the safety and efficacy of INT230-6 in patients with refractory or metastatic cancers. We completed the Phase 1 dose escalation portion of this study.
The Company has annually submitted safety data from all clinical trials to the FDA and Health Canada. Both regulatory agencies have reviewed the data and have permitted the Company to continue all clinical development programs without comment. The majority of drug related adverse events have been low grade (grade 1 or 2). As of October 20, 2021, a total of 11 patients out of 95 (12%) have had a grade 3 treatment related adverse event in study IT-01. The grade 3 events have been abdominal pain (4 patients), localized tumor pain (2 patients), fatigue (2 patients), and 1 case each of vomiting, dehydration and dizziness. There have been no grade 4 or 5 treatment related adverse events reported. We are currently conducting the Phase 2 portion of the trial, which consists of several different expansion cohorts. Four of the cohorts combine our product candidate with Merck’s Keytruda® (pembrolizumab) and 3 arms combine our drug candidate with Bristol-Myers Squibb’s drug Yervoy® (ipilimumab). We are also evaluating INT230-6 in a Phase 2 study (the INVINCIBLE study) in Canada as a treatment prior to surgery in early stage breast cancer.
1
Based on the broad range of data that we have generated from our preclinical experiments and clinical trials, we have observed that INT230-6 disperses widely throughout injected tumors, is absorbed well, penetrates and delivers the potent agents into tumor cells to kill them and activates a systemic immune response to fight the cancer. Our treatment approach utilizes intratumoral administration of INT230-6 to selectively induce tumor cell death and elicit an innate and adaptive anti-tumor immune response. Following injection of our product candidate, the tumors become highly necrotic, meaning that cancer cells die. After injection of INT230-6, tumors also become more amenable to immune cell infiltration. The tumor-killing process creates antigens, which are substances from the patient’s tumor that improve the recognition of the cancer by immune cells. While our product candidate is administered directly into the tumor, we have also observed in our preclinical studies and in our clinical trials that injections of INT230-6 can lead to a systemic immune response that attacks distal, uninjected tumors, a result known as an “abscopal” effect. Data generated in our trials show that our patented and patent pending drugs can extend life with less toxicity.
Between the metastatic study IT-01 and the INVINCIBLE study we have treated over 115 patients as of September 30, 2021.
Our Strengths
• Deep, Experienced Pharmaceutical Development, Finance and Accounting Management Team.
• Proprietary Drug Discovery platform, DfuseRxSM with Product Patent Protection in 37 Countries.
• Partnerships with World Leading Oncology Research Organizations and Major Pharmaceutical Companies.
• Clinical Data Demonstrates the Anti-cancer Activity in Humans in Multiple Cancers of Our Lead Product Candidate.
• Increased Survival observed in Metastatic Disease.
• Acceptable safety profile observed to date of the new drug/treatment approach.
• Fast Track Designation from FDA for INT230-6 in Triple Negative Breast Cancer.
• Phase 3 programs Designed and Planned.
• A Results-Oriented Organization.
• A Company Focused on Reaching the Market with its Lead Product Candidate.
Our Strategy
We seek to build a multi-product company that discovers, develops and commercializes tumor killing medicines that use novel diffusion mechanisms to penetrate cancer cells ushering in a fundamentally different methodology to treat cancer.
Key elements of our strategy include:
• Focus our resources to aggressively pursue the research and development of our novel medicine to transform patient lives.
• To always remember that taking care of and benefiting the patient is the most important element to being successful.
• Manage costs well by outsourcing research and development to qualified, academic, private or government laboratories to leverage the expertise while always maintaining our know-how, expertise and intellectual property.
• Build an internal team of experienced industry veterans that can work independently and who know how to get the product development job done.
• Create a large body of rigorous data, publications, presentations, collaborations and training materials about the new product candidates.
2
• Continuously find better methods to communicate to the medical community and patients of the power of our new approach.
• Continue our commitment to precision medicine and personalized care for each and every patient.
• Assure that our technology is fully understood, explored, and used as designed.
Summary of Risk Factors
Investing in our common stock involves significant risks. Any of the factors set forth in the section entitled “Risk Factors” may limit our ability to successfully execute our business strategy. You should carefully consider all of the information set forth in this prospectus and, in particular, you should evaluate the specific factors set forth in the section entitled “Risk Factors” in deciding whether to invest in our common stock. Some of the principal risks we face include:
• We are a clinical-stage biotechnology company with a limited operating history and have not generated any revenue to date from product sales.
• Since our inception, we have incurred, and for the foreseeable future anticipate that we will continue to incur, significant operating losses.
• The report of our independent registered public accounting firm for the year ended December 31, 2020 contains a statement with respect to substantial doubt as to our ability to continue as a going concern as a result of recurring losses from operations and negative cash flows.
• Even if we consummate this offering, we will need to raise substantial additional funding or we will be forced to delay, reduce or eliminate some of our product-development programs or commercialization efforts.
• We are largely dependent upon the success of our new intratumoral technology, which requires additional development and may never receive regulatory approval or be successfully commercialized.
• We have not completed clinical trials on any forms of cancer.
• Our prospects for obtaining additional financing are uncertain.
• The COVID-19 pandemic may affect our ability to initiate and complete current or future preclinical studies or clinical trials, disrupt regulatory activities or have other adverse effects on our business and operations.
• We have yet to obtain regulatory approval from the FDA, and therefore we are not currently permitted to market products made using our technology in the United States.
• Delays in FDA approval could be costly to us and prevent us from commercializing our product candidates effectively.
• Even if product candidates using our technology obtain approval, we will be subject to additional ongoing regulatory obligations and oversight.
• The FDA approval process is long, expensive and uncertain.
• Our ability to market a product may be limited by the uses that are approved for that product.
• We may be unable to export or sell products in foreign markets, which will limit our sales opportunities.
• We will rely on third parties to conduct preclinical research and any clinical trials.
• Third-party payors may not reimburse for the use of our product candidates or such reimbursement may be inadequate.
• We are dependent on third parties to manufacture components of the final drug products made using our technology.
• We purchase components for our product candidates from third parties, some of which may be sole-source suppliers.
3
• We have not entered into long term manufacturing and supply agreements with any producers.
• We have limited experience and may not be successful in commercializing products that use the Technology.
• Our plan to use collaborative arrangements with third parties to help finance and to market and sell products using our technology may not be successful.
• We will be dependent on healthcare professionals’ efforts to learn about our product candidates.
• We may need to establish clinical training and centers of excellence to educate and train physicians and healthcare payors, but the key opinion thought leadership required for initial market acceptance within the healthcare arena may take time to develop.
• Rapid technological developments in treatment methods for cancer and competition with other forms of cancer treatments could affect our ability to achieve meaningful revenues or profit.
• Our success depends in part on our ability to obtain patents, maintain trade secret protection, operate without infringing on the proprietary rights of third parties, and commercialize our technology prior to the expiration of our patent protection.
• We may be unable to protect our intellectual property rights because of our limited resources.
• We may be the subject of product liability claims or product recalls.
• If you purchase our common stock in this offering, you will incur immediate and substantial dilution in the book value of your shares.
4
Corporate Information
Intensity Therapeutics, Inc., a Delaware corporation, was incorporated on November 30, 2012, upon the conversion of its predecessor Intensity Therapeutics LLC. Our principal executive offices are located at 61 Wilton Road, 3rd Floor, Westport, CT 06880. Our telephone number at that location is (203) 221-7381. Our corporate website address is www.intensitytherapeutics.com. Information contained on, or that may be accessed through, our website is not incorporated by reference into this prospectus and should not be considered a part of this prospectus.
5
The Offering
|
Common stock offered by us |
2,142,858 shares. |
|
|
Option to purchase additional shares |
We have granted to the underwriters the option, exercisable for 45 days from the date of this prospectus, to purchase up to 321,428 additional shares of common stock at the initial public offering price, less estimated underwriting discounts and commissions. |
|
|
Common stock to be outstanding immediately |
|
|
|
Use of proceeds |
We estimate that the net proceeds from this offering will be approximately $13.1 million (or $15.1 million if the underwriters exercise in full their option to purchase additional shares of common stock), based on an assumed offering price of $7.00 per share (the mid-point of the price range set forth on the cover of this prospectus). |
|
|
We anticipate that we will use the net proceeds of this offering to advance and expand our clinical and preclinical development programs and for working capital and other general corporate purposes. For a more complete description of our intended use of the proceeds from this offering, see “Use of Proceeds.” |
||
|
Dividend policy |
We have no current plans to pay dividends on our common stock. See the section entitled “Dividend Policy” in this prospectus. |
|
|
Trading Symbol |
We have applied to list our common stock on Nasdaq under the symbol “INTS.” |
|
|
Risk factors |
You should read carefully the “Risk Factors” section of this prospectus for a discussion of factors that you should consider before deciding to invest in shares of our common stock. |
____________
(1) The number of shares of our common stock to be outstanding after this offering is based on 15,451,195 shares of common stock outstanding as of September 30, 2021, which includes 6,820,211 shares of our common stock outstanding as of September 30, 2021, plus 8,249,719 shares of our common stock issued upon the conversion of our preferred stock and 381,265 shares of our common stock that would be issued on the convertible note and accrued interest as of September 30, 2021 at a conversion price of $5.25 per share, and excludes:
• 1,822,500 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2021 under the 2013 Plan at a weighted average exercise price of $4.28 per share. Of these, 1,152,250 are exercisable at September 30, 2021 at a weighted average exercise price of $3.52 per share;
• 2,677,500 shares of our common stock reserved and available for future issuance under the 2013 Plan, as of September 30, 2021, which will cease to be available for issuance at the time that the 2021 Plan becomes effective;
• 646,500 shares of our common stock reserved and available for future issuance upon exercise of the outstanding warrants, as of September 30, 2021 at a weighted average exercise price of $3.00 per share. Of these, 568,974 are exercisable at September 30, 2021 at a weighted average price of $2.68 per share; and
• 3,000,000 shares of our common stock that will become available for future issuance under the 2021 Plan, which will become effective in connection with the completion of this offering.
Unless otherwise indicated, all information contained in this prospectus assumes no exercise by the underwriters of their option to purchase additional shares and no exercise of any other options or warrants.
6
Summary Financial Data
The following table sets forth a summary of our statement of comprehensive loss and summary of our balance sheet data for the periods indicated. Our historical results are not necessarily indicative of results that may be expected in the future. You should read the following summary financial data together with our financial statements and the related notes appearing elsewhere in this prospectus and the information in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
|
Statements of Operations Data: |
For the nine months ended |
For the years ended |
||||||||||||||
|
2021 |
2020 |
2020 |
2019 |
|||||||||||||
|
(Unaudited) |
(Unaudited) |
(Audited) |
(Audited) |
|||||||||||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
||||||||
|
Research and development costs |
$ |
4,419 |
|
$ |
3,644 |
|
$ |
5,050 |
|
$ |
4,437 |
|
||||
|
General and administrative costs |
|
1,184 |
|
|
922 |
|
|
1,173 |
|
|
1,238 |
|
||||
|
Total operating expenses |
|
5,603 |
|
|
4,566 |
|
|
6,223 |
|
|
5,675 |
|
||||
|
Loss from operations |
|
(5,603 |
) |
|
(4,566 |
) |
|
(6,223 |
) |
|
(5,675 |
) |
||||
|
Other income |
|
109 |
|
|
170 |
|
|
193 |
|
|
295 |
|
||||
|
Net loss |
$ |
(5,494 |
) |
$ |
(4,396 |
) |
$ |
(6,030 |
) |
$ |
(5,380 |
) |
||||
|
Loss per share, basic and diluted common |
$ |
(0.81 |
) |
$ |
(0.64 |
) |
$ |
(0.88 |
) |
$ |
(0.79 |
) |
||||
|
Weighted average number of common stock, basic and diluted |
|
6,820,211 |
|
|
6,818,631 |
|
|
6,819,026 |
|
|
6,805,994 |
|
||||
|
Balance Sheet Data: |
As of September 30, |
As of December 31, |
||||||||||||||
|
2021 |
2020 |
2020 |
2019 |
|||||||||||||
|
(Unaudited) |
(Unaudited) |
|||||||||||||||
|
Cash and cash equivalents |
$ |
7,408 |
|
$ |
3,137 |
|
$ |
9,316 |
|
$ |
3,829 |
|
||||
|
Total current assets |
|
7,698 |
|
|
11,353 |
|
|
9,628 |
|
|
8,649 |
|
||||
|
Total assets |
|
8,109 |
|
|
11,562 |
|
|
10,150 |
|
|
8,931 |
|
||||
|
Total liabilities |
|
4,648 |
|
|
1,604 |
|
|
1,708 |
|
|
1,307 |
|
||||
|
Redeemable convertible preferred stock |
|
10,000 |
|
|
10,000 |
|
|
10,000 |
|
|
10,000 |
|
||||
|
Total deficiency |
$ |
(6,539 |
) |
$ |
(42 |
) |
$ |
(1,558 |
) |
$ |
(2,376 |
) |
||||
7
You should carefully consider the risks described below before buying shares in Intensity Therapeutics, Inc. These are risks and uncertainties that management believes are most likely to be material and therefore are important for an investor to consider. Our business operations and results may also be adversely affected by additional risks and uncertainties not presently known to us, or which are currently deemed immaterial, or which are similar to those faced by other companies in the pharmaceutical industry or business in general. If any of the following risks or uncertainties actually occurs, our business, financial condition, results of operations, or cash flows would likely suffer. In that event, the value of our stock could decline, perhaps significantly.
Risks Related to Our Business, Financial, and Investment Conditions
We are a clinical-stage biotechnology company with a limited operating history and have not generated any revenue to date from product sales.
We are a clinical-stage, pre-commercial company with only a limited operating history upon which to base an evaluation of our current business and future prospects and how we will respond to competitive, financial or technological challenges. Biotechnology product development is a highly speculative undertaking and involves a substantial degree of risk. We were incorporated under the laws of the State of Delaware in November 2012. Since inception, we have focused substantially all of our efforts and financial resources on raising capital and developing our initial product candidates. We have no products approved for commercial sale and therefore have never generated any revenue from product sales, and we do not expect to do so in the foreseeable future. We have not obtained regulatory approvals for any of our product candidates. Consequently, the revenue-generating potential of our business is unproven and uncertain. Even if our product candidates receive regulatory approval, we may be unable to successfully introduce and market them at prices that would permit us to operate profitably.
We have incurred significant operating losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
To date, we have financed our operations primarily through an initial investment from our founder and the issuance and sale of common stock, our convertible preferred stock and convertible debt notes, to outside investors in private equity financings. From our inception through September 30, 2021, we raised an aggregate of $32.1 million of gross proceeds from such transactions. As of September 30, 2021, our cash and cash equivalents and investments were $7.4 million. We have incurred net losses in each year since our inception, and we had an accumulated deficit of $28.7 million as of September 30, 2021. For the nine months ended September 30, 2021 and for the years ended December 31, 2020 and December 31, 2019, we reported net losses of $5.5 million, $6.0 million and $5.4 million, respectively. The report of our independent registered public accounting firm for the year ended December 31, 2020 included herein contains an explanatory paragraph indicating that there is substantial doubt as to our ability to continue as a going concern as a result of recurring losses from operations and negative cash flows.
We expect to continue to incur significant expenses and operating losses over the next several years and for the foreseeable future. Substantially all of our operating losses have resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations. We expect our research and development expenses to significantly increase in connection with the commencement and continuation of clinical trials of our product candidates. In addition, if we obtain marketing approval for our product candidates, we will incur significant sales, marketing and manufacturing expenses. Once we are a public company, we will incur additional costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing biotechnology products, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do become profitable, we may not be able to sustain or increase our profitability on a quarterly or annual basis. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ deficit and working capital.
8
If we fail to establish and maintain an effective system of internal control, we may not be able to report our financial results accurately or to prevent fraud. Any inability to report and file our financial results accurately and timely could harm our reputation and adversely impact the future trading price of our common stock.
Effective internal control is necessary for us to provide reliable financial reports and prevent fraud. However, because of our limited resources, there are limited controls over information processing. We have a material weakness due to a lack of segregation of duties, since we have a limited administrative staff. Our management is composed of a small number of individuals resulting in a situation where limitations on segregation of duties exist. We have focused our segregation of duties to ensure that the actual payments are performed separately from the accounting staff, and the Chief Executive Officer performs a robust review of the financial statements on a monthly basis. All accounting entries and the creation of financial statements, however, are performed by a single person. To remedy this situation, we would need to hire additional staff. In August 2021, we hired a Chief Financial Officer to add a layer of supervision and control. Currently, we are unable to hire additional staff to facilitate greater segregation of duties but will reassess its capabilities after completion of the Offering.
Our small size and internal control deficiencies may adversely affect our financial condition, results of operation and access to capital. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed.
The report by our auditors includes a paragraph that states that substantial doubt exists about the Company’s ability to continue as a going concern.
The report of our independent registered public accounting firm for the year ended December 31, 2020 included herein contains an explanatory paragraph indicating that there is substantial doubt as to our ability to continue as a going concern as a result of recurring losses from operations and negative cash flows. We do not have a history of earnings and, as a result, substantial doubt exists about our ability to continue as a going concern. Further, without the proceeds of this offering, we do not have sufficient cash to continue with our business plan for the next 12 months. Also, at any time on or after May 18, 2022, the holders of at least two-thirds of the then outstanding shares of Series A Preferred Stock may elect to cause the Company to redeem all, but not less than all, of the shares of Series A Preferred Stock at a redemption price per share of $2.00, or $10,000,000 in total.
Our continued operations are dependent on our ability to complete equity or debt financings or generate profitable operations. Such financings may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we are unable to obtain adequate funding from this proposed offering or in the future, or if we are unable to generate revenue to achieve and sustain profitability, we may not be able to continue as a going concern. We believe that there is substantial doubt as to whether we can raise sufficient funding in order for us to continue operations.
Even if we consummate this offering, we will need to raise substantial additional funding. If we are unable to raise capital when needed, we would be forced to delay, reduce or eliminate some or all of our product development programs or commercialization efforts.
The development of biotechnology products is capital-intensive and we expect our expenses to significantly increase in connection with our ongoing activities, particularly as we continue our ongoing clinical trials or initiate future trials and pursue the research and development of, and seek marketing approval for, our product candidates. Our future capital requirements will depend on and could increase significantly as a result of many factors, including:
• our research and product development programs, including clinical studies;
• the timing and costs of our various U.S. and foreign regulatory filings, obtaining approvals, and complying with regulations;
• the timing and costs associated with developing manufacturing operations;
• the timing of product commercialization activities, including marketing and distribution arrangements;
• the timing and costs involved in preparing, filing, prosecuting, defending, and enforcing intellectual property rights; and
• the impact of competing technological and market developments.
9
We expect that the net proceeds from this offering, together with our existing cash and cash equivalents and investments will be sufficient to fund our operations and capital expenditure requirements through September 2023. Accordingly, we will need to obtain substantial additional funding to continue our operations. We cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborators or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects. Any additional fundraising efforts may also divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate certain of our research and development programs or future commercialization efforts, and may be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
We are largely dependent upon the success of our new intratumoral technology, which will require additional development before we may be able to seek regulatory approval and may never receive regulatory approval or be successfully commercialized.
The Intensity Therapeutics Technology, a platform for the creation of products to improve treatment of cancer patients, is our only technology. Our entire focus has been on developing, commercializing, and ultimately obtaining regulatory authorizations and approvals of product candidates using this technology. We have invested, and we expect to continue to invest, significant efforts and financial resources in its development. Our ability to generate meaningful revenue, which may not occur for the foreseeable future, if ever, will depend heavily on the successful development, regulatory approval and commercialization of our technology. If we are unable to develop the Intensity Therapeutics Technology, obtain regulatory approval, and sell products using the technology, we will not generate operating revenue or become profitable, and we may be forced to terminate or cease operations.
We have not completed clinical trials on any forms of cancer, and we are subject to risks and challenges that may prevent or delay the completion of our clinical trials.
We have only two clinical trials in progress. One on-going study is a multi-cohort clinical trial testing our product candidate alone or combined with Keytruda® or with Yervoy®. The other study is a randomized Phase 2 study in presurgical breast cancer. Our program is in the early stage. Only 115 patients have been dosed in our clinical trials as of September 30, 2021. We have not demonstrated any survival benefit in a statistically significant and meaningful manner. We have not demonstrated sufficient safety of any product candidate for FDA approval. Our largest dose on any given day so far has been 244mL containing 122 mg of cisplatin and 24.4 mg of vinblastine sulfate. We have no indication that higher doses or any dose will be safe or effective.
We intend to conduct clinical trials for multiple indications, and it may take several years to complete the testing of our product candidates and technology for the indications for which we wish to obtain approval. Failure or delay can occur at any stage of development, for many reasons, including:
• any pre-clinical or clinical test may fail to produce results satisfactory to the FDA or foreign regulatory authorities and preclude us from testing in humans;
• pre-clinical or clinical data can be interpreted in different ways, which could delay, limit, or prevent regulatory approval;
• negative or inconclusive results from a pre-clinical study or clinical trial or adverse medical events during a clinical trial could cause a pre-clinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful;
10
• the FDA or foreign regulatory authorities can place a clinical hold on a trial if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury;
• changes in regulatory agency policies during the period in which we are developing a system, or the period required for review of any application for regulatory agency approval;
• our clinical trials may not demonstrate the safety and efficacy of any system or result in marketable products;
• the FDA or foreign regulatory authorities may request additional clinical trials, including more than one Phase 3 trial, relating to any potential NDA submissions;
• the FDA or foreign regulatory authorities may change their approval policies or adopt new regulations that may negatively affect or delay our ability to bring a system to market or require additional clinical trials; and
• a system may not be approved for all the requested indications.
We face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
The biopharmaceutical industry is characterized by intense competition and rapid innovation. We face competition from major pharmaceutical, specialty pharmaceutical and biotechnology companies among others with respect to INT230-6 and will face similar competition with respect to any product candidates that we may seek to develop or commercialize in the future. We compete in pharmaceutical, biotechnology and other related markets that develop immune-oncology therapies for the treatment of cancer. There are other companies working to develop new drugs, immunotherapies and other approaches for the treatment of cancer including divisions of large pharmaceutical and biotechnology companies of various sizes. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations and well-established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly as they develop novel approaches to treating disease indications that our product candidates are also focused on treating. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel therapeutics or to in-license novel therapeutics that could make the product candidates that we develop obsolete. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors, either alone or with collaborative partners, may succeed in developing, acquiring or licensing on an exclusive basis drug or biologic products that are more effective, safer, more easily commercialized or less costly than our product candidates or may develop proprietary technologies or secure patent protection that we may need for the development of our technologies and products. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety, tolerability, reliability, convenience of use, price and reimbursement.
There are a number of companies trying to develop intratumoral therapies. However, most of our competitors are currently focused on intratumoral treatment approaches that stimulate immune cells to achieve inflammation rather than directly killing a tumor. This shift to a pure immune-oncology (IO) treatment has reopened the investigations into intratumoral approaches focusing on activating local immune response. Amgen markets a novel genetically modified oncolytic viral-based immunotherapeutic, talimogene laherparepvec (T-Vec), that has been approved for IT use in cutaneous melanoma. While T-Vec is approved solely for local treatment of localized cutaneous melanoma, the drug has not been shown to improve overall survival or have any effect on distal metastases, which will be a critical factor to broader use. Another viral based system is being developed by Replimune. RP1 is Replimune’s genetically modified herpes simplex type 1 virus that is designed to directly destroy tumors and to generate an anti-tumor immune response. This product is being evaluated in a Phase 1/2, open label, multicenter, dose escalation and expansion, first-in-human (FIH) clinical study to evaluate the safety and tolerability, biodistribution, shedding, and preliminary efficacy of RP1 alone and in combination with nivolumab in adult subjects with advanced and/or refractory solid tumors. The IGNYTE Study, which started in 2017, includes a dose escalation phase for single agent RP1, an expansion phase with
11
a combination of RP1 and nivolumab and a Phase 2 portion in specified tumor types for the combination therapy. Dose escalation of RP1 by intratumoral injection in superficial tumors and in visceral tumors. The objective of this viral approach is to transfect the granulocyte-macrophage colony-stimulating factor gene into the tumor microenvironment to recruit a local inflammatory response that would promote a systemic immune response.
Oncosec Immunotherapies Inc. is developing cytokine-based intratumoral immunotherapies to stimulate the body’s immune system to target and attack cancer. The Company built a clinical pipeline utilizing their primary technology, TAVO™ (tavokinogene telseplasmid), as a potential treatment for multiple cancer indications either as a monotherapy or in combination with leading checkpoint inhibitors. TAVO is DNA-based interleukin-12 (IL-12), a naturally occurring protein in the body with immune-stimulating functions. TAVO is administered directly into the tumor using the Company’s proprietary electroporation (EP) gene delivery system, which employs a series of momentary energy pulses. Those pulses are designed to increase the permeability of the cell membrane and facilitate uptake of IL-12 coded DNA into cells.
Other local treatment approaches being explored by companies such as Merck also attempt to recruit the immune system cells into the local tumor microenvironment with intratumoral delivery of other agents. Data on several other intratumorally-delivered agents such as STING agonists, RIG-1, and TLR9 have been presented at major cancer conferences.
Our belief is that our competitors have formulated their products without consideration of the inability of water-based products to be well absorbed into a tumor’s lipophilic, high-pressure microenvironment. Attempts at the stimulation of an inflammatory response or efforts to attract immune cells into a hostile live, rapidly growing tumor still pose a number of challenges. Accordingly, there remains a continued unmet need for the development of direct IT therapies for solid tumors that provide high local killing efficacy coupled with nontoxic systemic anti-cancer effects. We believe we have created a product candidate with the necessary chemistry to overcome the local delivery challenges. Evidence shows the mechanism of tumor killing achieved by our drug candidate also leads to systemic immune activation in certain cancers.
We anticipate competing with other companies that are focused on treating disease indications that our product candidates are also focused on treating. A competitor may develop technologies focused on the same disease pathway as our technology or may focus on treating the targeted disease in a completely different manner. To the extent a new drug is developed that is more efficacious than any product candidate developed by us, this could reduce or negate the need for our product candidate. In addition, while we believe our product candidates may be used in conjunction with existing or emerging standard of care (SOC) in certain disease indications, as companies continue to improve upon existing standard of care, more efficacious drug therapies could become available, reducing or completely negating the benefit of our product candidates. Our competitors may also include companies that are or will be developing therapies for the same therapeutic areas that we are targeting within our early pipeline.
Even if we are successful in achieving regulatory approval to commercialize a product candidate ahead of our competitors, our future pharmaceutical products may face direct competition from generic and other follow-on drug products. Any of our product candidates that may achieve regulatory approval in the future may face competition from generic products earlier or more aggressively than anticipated, depending upon how well such approved products perform in the U.S. prescription drug market. Our ability to compete also may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products. Generic products are expected to become available over the coming years. Even if our product candidates achieve marketing approval, they may be priced at a significant premium over competitive generic products, if any have been approved by then.
In addition to creating the 505(b)(2) NDA pathway, the Hatch-Waxman Amendments to the federal Food, Drug, and Cosmetic Act (FDCA) authorized the FDA to approve generic drugs that are the same as drugs previously approved for marketing under the NDA provisions of the statute pursuant to ANDAs. An ANDA relies on the preclinical and clinical testing conducted for a previously approved reference listed drug (“RLD”), and must demonstrate to the FDA that the generic drug product is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug and also that it is “bioequivalent” to the RLD. The FDA is prohibited by statute from approving an ANDA when certain marketing or data exclusivity protections apply to the RLD. If any such competitor or third party is able to demonstrate bioequivalence without infringing our patents, then this competitor or third party may then be able to introduce a competing generic product onto the market.
12
We cannot predict the interest of potential follow-on competitors or how quickly others may seek to come to market with competing products, whether approved as a direct ANDA competitor or as a 505(b)(2) NDA referencing one of our future drug products. If the FDA approves generic versions of our drug candidates in the future, should they be approved for commercial marketing, such competitive products may be able to immediately compete with us in each indication for which our product candidates may have received approval, which could negatively impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on our investments in those product candidates.
Even if we obtain regulatory approval of our product candidates, the availability and price of our competitors’ products could limit the demand and the price we are able to charge for our product candidates. We may not be able to implement our business plan if the acceptance of our product candidates is inhibited by price competition or the reluctance of physicians to switch from existing methods of treatment to our product candidates, or if physicians switch to other new drug or biologic products or choose to reserve our product candidates for use in limited circumstances. For additional information regarding our competition, see “Business — Competition.”
The COVID-19 pandemic has spread worldwide and may affect our ability to initiate and complete current or future preclinical studies or clinical trials, disrupt regulatory activities or have other adverse effects on our business and operations. In addition, this pandemic has caused substantial disruption in the financial markets and may adversely impact economies worldwide, both of which could result in adverse effects on our business and operations.
The COVID-19 pandemic, which began in December 2019 and has spread worldwide, has caused many governments to implement measures to slow the spread of the outbreak through quarantines, travel restrictions, heightened border scrutiny, and other measures. The outbreak and government measures taken in response have also had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. The future progression of the outbreak and its effects on our business and operations are uncertain. We and our contract manufacturing organizations or clinical sites, or CMOs, and contract research organizations, or CROs, may face disruptions that may affect our ability to initiate and complete preclinical studies or clinical trials or raise capital to finance our business.
Our prospects for obtaining additional financing, as needed, are uncertain and our failure to obtain needed financing could affect our ability to pursue future growth.
Even if this offering is successful, we will need to raise additional funds in the future to develop or enhance our product candidates, to fund expansion, to conduct additional clinical trials and to fund general operating expenses. There is no assurance that additional financing will be available on terms favorable to us, or at all. If additional funds are raised through the issuance of equity or convertible debt securities, the percentage ownership of our stockholders would be reduced, and these securities might have rights, preferences, or privileges senior to those of our current stockholders. If adequate funds are not available on acceptable terms, our ability to fund our expansion, take advantage of unanticipated opportunities, develop or enhance services or products, or otherwise respond to competitive pressures would be significantly limited.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new products to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical employees and stop critical activities.
13
Separately, the FDA has announced its commitment to achieving timely reviews of applications for medical products during the COVID-19 pandemic in line with its user fee performance goals; however, the FDA may not be able to continue its current pace and review timelines could be extended, including where a pre-approval inspection or an inspection of clinical sites is required and due to the COVID-19 pandemic and travel restrictions FDA is unable to complete such required inspections during the review period. On March 10, 2020, the FDA announced its intention to postpone most inspections of foreign manufacturing facilities, and on March 18, 2020, the FDA temporarily postponed routine surveillance inspections of domestic manufacturing facilities. Subsequently, on July 10, 2020, the FDA announced its intention to resume certain on-site inspections of domestic manufacturing facilities subject to a risk-based prioritization system. The FDA intends to use this risk-based assessment system to identify the categories of regulatory activity that can occur within a given geographic area, ranging from mission critical inspections to resumption of all regulatory activities. Additionally, on April 15, 2021, the FDA issued a guidance document in which the FDA described its plans to conduct voluntary remote interactive evaluations of certain drug manufacturing facilities and clinical research sites. According to the guidance, the FDA intends to request such remote interactive evaluations in situations where an in-person inspection would not be prioritized or deemed mission-critical, or where direct inspection is otherwise limited by travel restrictions, but where the FDA determines that remote evaluation would still be appropriate. Regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic and may experience delays in their regulatory activities. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, upon completion of this offering and in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Risks Related to FDA and Foreign Regulatory Approval
Clinical development involves a lengthy, complex and expensive process, with an uncertain outcome, and the results of preclinical studies and early-stage clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials.
The development and approval process in the United States may take many years, require substantial resources, and may never lead to the approval of any of our product candidates by the FDA for use in the United States. To obtain the requisite regulatory approvals to commercialize any product candidates, we must demonstrate through extensive preclinical studies and clinical trials that our product candidates are safe and effective in humans. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. In particular, the general approach for FDA approval of a new drug is dispositive data from one or two adequate and well-controlled, Phase 3 clinical trials of the relevant drug in the relevant patient population. Phase 3 clinical trials typically involve hundreds of patients, have significant costs and take years to complete. A product candidate can fail at any stage of testing, even after observing promising signals of activity in earlier preclinical studies or clinical trials. The results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. In addition, initial success in clinical trials may not be indicative of results obtained when such trials are completed. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biotechnology and biopharmaceutical industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. Most product candidates that commence clinical trials are never approved as therapeutic products, and there can be no assurance that any of our future clinical trials will ultimately be successful or support further clinical development of INT230-6 or any of our other product candidates. Product candidates that appear promising in the early phases of development may fail to reach the market for several reasons, including:
• preclinical studies or clinical trials may show the product candidates to be less effective than expected (e.g., a clinical trial could fail to meet its primary endpoint(s)) or to have unacceptable side effects or toxicities;
• failure to establish clinical endpoints that applicable regulatory authorities would consider clinically meaningful;
14
• failure to receive the necessary regulatory approvals;
• manufacturing costs, formulation issues, pricing or reimbursement issues, or other factors that make a product candidate uneconomical; and
• the proprietary rights of others and their competing products and technologies that may prevent one of our product candidates from being commercialized.
In addition, differences in trial design between early-stage clinical trials and later-stage clinical trials make it difficult to extrapolate the results of earlier clinical trials to later clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in clinical trials have nonetheless failed to obtain marketing approval of their products.
Additionally, we expect that some of our trials will be open-label studies, where both the patient and investigator know whether the patient is receiving the investigational product candidate as a monotherapy or in combination with an existing approved drug. Most typically, open-label clinical trials test only the investigational product candidate and sometimes do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge. Therefore, it is possible that positive results observed in open-label trials will not be replicated in later placebo-controlled trials.
In addition, the standards that the FDA and comparable foreign regulatory authorities use when regulating our product candidates require judgment and can change, which makes it difficult to predict with certainty how they will be applied. Although we are initially focusing our efforts on development of small-molecule drug products, we may in the future pursue development of biological products, which could make us subject to additional regulatory requirements. Any analysis we perform of data from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations. Examples of such regulations include future legislation or administrative action, or changes in FDA policy during the period of product development and FDA regulatory review. We cannot predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be. The FDA may also require a panel of experts, referred to as an Advisory Committee, to deliberate on the adequacy of the safety and efficacy data to support approval. The opinion of the Advisory Committee, although not binding, may have a significant impact on our ability to obtain approval of any product candidates that we develop.
We may seek to conduct clinical trials in foreign countries, as well as in the United States. If we continue to seek to conduct clinical trials in foreign countries or pursue marketing approvals in foreign jurisdictions, we must comply with numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process varies among countries and may include all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Moreover, the time required to obtain approval from foreign regulatory agencies may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities outside the United States and vice versa.
Successful completion of clinical trials is a prerequisite to submitting a marketing application to the FDA and similar marketing applications to comparable foreign regulatory authorities, for each product candidate and, consequently, the ultimate approval and commercial marketing of any product candidates. We may experience negative or inconclusive results, which may result in our deciding, or our being required by regulators, to conduct additional clinical studies or trials or abandon some or all of our product development programs, which could have a material adverse effect on our business.
We will likely need separate regulatory approvals for every therapeutic agent or combination of compounds that we intend to develop and market using our technology.
Although many drugs have been approved by the FDA for use as therapeutic agents, regulatory approval is likely required in the United States for the combined enhancer component with the drug component(s) and the specific indication, dose, and route of administration of the therapeutic agent or agents used in our system.
15
We will likely need to obtain separate regulatory approvals for products using our technology with every therapeutic agent or combination of compounds used with our system that we intend to market. All the manufacturing facilities used to manufacture components or assemble our system must be inspected and meet legal requirements. Securing regulatory approval requires the submission of extensive pre-clinical and clinical data and other supporting information for each proposed therapeutic indication to establish to the FDA’s satisfaction the product’s safety, efficacy, potency, and purity for each intended use. The pre-clinical testing and clinical trials of any products using our technology with any therapeutic agent or compound we use must comply with the regulations of the FDA and other federal, state, and local government authorities in the United States. Clinical development is a long, expensive, and uncertain process and is subject to delays. We may encounter delays or rejections for various reasons, including our inability to enroll enough patients to complete our clinical trials. Moreover, approval policies or regulations may change. If we do not obtain and maintain regulatory approval for our system and our use of therapeutic agents, our results of operations will be harmed.
Failure to obtain, or delay in obtaining, regulatory approvals would likely have a material adverse effect on our business, financial condition and results of operations.
During its development, our product candidates and technology will be subject to extensive and rigorous government regulation by the U.S. Food and Drug Administration (FDA) and possibly other foreign regulatory agencies. The FDA regulates the research, development, pre-clinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of pharmaceutical and medical device products. Failure to comply with FDA and other applicable regulatory requirements, either before or after product approval, may subject us to administrative or judicially imposed sanctions.
We are not permitted to market products made using our technology in the United States unless and until we obtain regulatory approval from the FDA.
To market the product candidate in the United States, we must submit to the FDA and obtain FDA approval of a New Drug Application (NDA). An investigational new drug (IND) application is the first step in the regulatory process. Under an IND, a Company develops a drug in the hopes of someday submitting to FDA the NDA to permit marketing of the drug. An NDA must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing, and controls (CMC) to demonstrate the safety and effectiveness of the applicable product candidate. Regulatory approval of an NDA is not guaranteed. The number and types of preclinical studies and clinical trials that will be required varies depending on the product candidate, the disease or condition that the product candidate is designed to target, and the regulations applicable to any product candidate. Despite the time and expense associated with preclinical and clinical studies, failure can occur at any stage and we could encounter problems that cause us to repeat or perform additional preclinical studies, CMC studies or clinical trials. The FDA and similar foreign authorities could delay, limit or deny approval of a product candidate for many reasons, including because they:
• may not deem a product candidate to be adequately safe and effective;
• may not find the data from preclinical studies, CMC studies, and clinical trials to be sufficient to support a claim of safety and efficacy;
• may interpret data from preclinical studies, CMC studies, and clinical trials significantly differently than we do;
• may not approve the manufacturing processes or facilities associated with our product candidates;
• may change approval policies (including with respect to our product candidates’ class of drugs) or adopt new regulations; or
• may not accept a submission due to, among other reasons, the content or formatting of the submission.
Delays in FDA approval could be costly to us and prevent us from commercializing our product candidates effectively.
The regulatory review and approval process is lengthy, expensive, and inherently uncertain. As part of the U.S. Prescription Drug User Fee Act, the FDA has a goal to review and act on a percentage of all submissions in a given time frame. The general review goal for a drug application is ten to twelve months for a standard application and six months for a priority review application. The FDA’s review goals are subject to change and it is unknown whether the review of an NDA filing for any of our product candidates will be completed within the FDA’s review goals or
16
will be delayed. Moreover, the duration of the FDA’s review may depend on the number and types of other NDAs that are submitted to the FDA around the same time. The development and approval process may take many years, require substantial resources, and may never lead to the approval of a product. Failure to obtain or delays in obtaining regulatory approvals may:
• adversely affect the commercialization of our current technology or any products that we develop in the future;
• impose additional costs on us;
• diminish any competitive advantages that may be attained; and
• adversely affect our ability to generate revenues.
We have received, and may continue to seek, Breakthrough Therapy Designation or Fast Track Designation from the FDA, for certain of our product candidates, but receipt of either such designation may not actually lead to a faster development or regulatory review or approval process.
In 2018, we received Fast Track Designation by the FDA to use INT230-6 in metastatic triple negative breast cancer for patients whose cancer has progressed following one or two prior drug treatments. We may continue to seek Breakthrough Therapy Designation or Fast Track Designation for our product candidates or for other indications.
A breakthrough therapy is defined as a product that is intended, alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For products that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Products designated as breakthrough therapies by the FDA can also be eligible for accelerated approval.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough Therapy Designation for a product candidate may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the products no longer meet the conditions for qualification and rescind the breakthrough designation.
If a product is intended for the treatment of a serious or life-threatening condition and the product demonstrates the potential to address unmet medical needs for this condition, the product sponsor may apply for Fast Track Designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular product candidate is eligible for this designation, we cannot assure you that the FDA would decide to grant it. Even though we have received Fast Track Designation to use INT230-6 in certain indications, or if we receive Fast Track Designation for other drug products or indications, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons. The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. The enrollment of patients depends on many factors, including:
• the patient eligibility and exclusion criteria defined in the protocol;
• the size of the patient population required for analysis of the trial’s primary endpoints;
17
• the proximity of patients to trial sites;
• the design of the trial;
• our ability to recruit clinical trial investigators with the appropriate competencies and experience;
• our ability to obtain and maintain patient consents; and
• the risk that patients enrolled in clinical trials will drop out of the trials before completion.
In addition, our clinical trials will compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition will reduce the number and types of patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigators is limited, we expect to conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials in such clinical trial site.
Delays in patient enrollment may result in increased costs or may affect the timing or outcome of our future clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our product candidates.
We will rely on third parties to conduct certain of the preclinical research and any clinical trials for products using our technology, and if those third parties perform in an unsatisfactory manner, it may harm our business.
We do not currently have the ability to independently conduct any clinical trials. We intend to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our preclinical studies and clinical trials, and we expect to have limited influence over their actual performance. We rely upon CROs to monitor and manage data for our clinical programs, as well as the execution of future preclinical studies. We expect to control only certain aspects of our CROs’ activities. Nevertheless, we will be responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
We and our CROs are required to comply with the good laboratory practices, or GLPs, and GCPs, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities in the form of International Conference on Harmonization guidelines for any of our product candidates that are in preclinical and clinical development. The regulatory authorities enforce GCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. Although we rely on CROs to conduct GCP-compliant clinical trials, we remain responsible for ensuring that each of our GLP preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol and applicable laws and regulations. If we or our CROs fail to comply with GCPs, the clinical data generated in our clinical trials may be deemed unreliable, and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. Accordingly, if our CROs fail to comply with these regulations or fail to recruit a sufficient number of subjects, we may be required to repeat clinical trials, which would delay the regulatory approval process.
Our reliance on third parties to conduct clinical trials will result in less direct control over the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with CROs and other third parties can be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Such parties may:
• have staffing difficulties;
• fail to comply with contractual obligations;
• experience regulatory compliance issues; or
• undergo changes in priorities or become financially distressed.
These factors may adversely affect the willingness or ability of third parties to conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or fail to comply with regulatory requirements,
18
or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for any other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, any product candidate that we develop. As a result, our financial results and the commercial prospects for any product candidate that we develop would be harmed, our costs could increase, and our ability to generate revenue could be delayed. While we will have agreements governing their activities, our CROs will not be our employees, and we will not control whether or not they devote sufficient time and resources to our future clinical and preclinical programs. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials, or other drug development activities which could harm our business. We face the risk of potential unauthorized disclosure or misappropriation of our intellectual property by CROs, which may reduce our trade secret protection and allow our potential competitors to access and exploit our proprietary technology.
If our relationship with any of these CROs terminates, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or adding additional CROs involves substantial cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can negatively impact our ability to meet our desired clinical development timelines. While we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a negative impact on our business, financial condition and prospects.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval of our product candidates.
Even if products using our technology are approved by the FDA or any other regulatory agency, we will be subject to additional ongoing regulatory obligations and oversight in the U.S. and other countries where we obtain approval.
For example, we may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. In addition, if the FDA approves a product candidate, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, and continued compliance with FDA Current Good Manufacturing Processes (CGMPs), good clinical practices (GCPs), and good laboratory practices, which are regulations and guidelines enforced by the FDA for all products in clinical development and for any clinical trials that we conduct post-approval. In addition, post-marketing requirements for our product candidates may include implementation of a Risk Evaluation and Mitigation Strategies (REMS) to ensure that the benefits of the product outweigh its risks. A REMS may include a Medication Guide, a patient package insert, a communication plan to healthcare professionals, and/or other elements to assure safe use of the product. Compliance with all these requirements, and any other requirements imposed upon us by U.S. or overseas regulators, could be costly to us, and failure to comply with these requirements could cause us to lose any marketing approval that we may have obtained, subject us to sanctions and jeopardize our ability to commercialize our product candidates.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with any third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• refusals or delays in the approval of applications or supplements to approved applications;
• refusal of a regulatory authority to review pending market approval applications or supplements to approved applications;
• restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market or voluntary or mandatory product recalls or seizures;
19
• fines, warning letters, or holds on clinical trials;
• import or export restrictions;
• injunctions or the imposition of civil or criminal penalties;
• restrictions on product administration, requirements for additional clinical trials, or changes to product labeling or REMS programs; or
• recommendations by regulatory authorities against entering into governmental contracts with us.
Even if we obtain regulatory approval for our product candidates using our technology in the United States, our ability to market a product would be limited to those uses that are approved for that product.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities, and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. In the United States, we intend to seek approval for products for various types of cancer. If the FDA approves any drug application, our ability to market and promote a product would be limited to the indication tested for a specific disease, so even with FDA approval, products using our technology may only be promoted in this limited market. Physicians may prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, including oncology. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding promotion of approved drug products for off-label use, and FDA approval may otherwise limit our sales practices and our ability to promote, sell, and distribute a product. Thus, we may only market products using our technology, if approved by the FDA, for its approved indication and we could be subject to enforcement action for off-label marketing.
Further, if there are any modifications to an approved product, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in regulatory enforcement actions and adverse publicity.
If future clinical trials are unsuccessful, significantly delayed or not completed, we may not be able to market products for other indications or our technology.
If we do not obtain required approvals in other countries in which we aim to market our product candidates, we will not be able to export or sell the products in those markets, which will limit our sales opportunities.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, while a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, similar foreign regulatory authorities must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval and licensure procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions.
Our lack of experience conducting clinical trials outside the United States and Canada may negatively impact the approval process in foreign countries where we intend to seek approval for the products using our technology. We have not previously conducted multi-national clinical trials.
If we are unable to obtain and maintain required approval from one or more foreign jurisdictions where we would like to sell products using our technology, we will be unable to market products as intended, our international market opportunity will be limited, and our results of operations will be harmed.
20
If no product candidates using our technology are approved by the FDA or other regulatory body, third-party payors in the United States or anywhere will not reimburse the use of our product candidates. Even if approval is obtained, inadequate reimbursement may harm results of operations.
Following regulatory approval, we intend to seek reimbursement by third-party payors for the products created by our technology. There are no assurances that third-party payors in the United States or other countries will agree to cover the cost of products using our technology at all or at rates that are adequate to cover actual costs. Further, third-party payors may deny reimbursement if they determine that our product candidates are not used in accordance with established payor protocols regarding cost effective treatment methods or are used outside their approved indication or for forms of cancer not specifically approved by the FDA or other foreign regulatory bodies in the future. Without reimbursement, physicians, hospitals, and other healthcare providers may be less likely to prescribe our product candidates thereby harming our results of operations. Without adequate reimbursement, we may not be able to successfully commercialize systems.
Risks Related to Manufacturing, Commercialization, and Market Acceptance of Products made using our Technology
We intend to rely on third parties to produce clinical and commercial supplies of our product candidates.
We do not own or operate facilities for drug manufacturing, storage and distribution, or testing. We are dependent on third parties to manufacture the clinical supplies of our current and any future product candidates. The facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA pursuant to inspections that will be conducted after we submit our NDA to the FDA. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the cGMP requirements, for manufacture of both active drug substance and finished drug product. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, we will not be able to secure and/or maintain regulatory approval for our product candidates. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Any significant delay in the supply of a product candidate, or the raw material components thereof, for an ongoing clinical trial due to the need to replace a third-party manufacturer could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates.
We also intend to rely on third-party manufacturers to supply us with sufficient quantities of our product candidates to be used, if approved, for commercialization. We do not yet have a commercial supply agreement for commercial quantities of drug substance or drug product. If we are not able to meet market demand for any approved product, it would negatively impact our ability to generate revenue, harm our reputation, and could have an adverse effect on our business and financial condition.
Further, our reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates ourselves, including:
• inability to meet our product specifications and quality requirements consistently;
• delay or inability to procure or expand sufficient manufacturing capacity;
• issues related to scale-up of manufacturing;
• costs and validation of new equipment and facilities required for scale-up;
• our third-party manufacturers may not be able to execute our manufacturing procedures and other logistical support requirements appropriately;
• our third-party manufacturers may fail to comply with cGMP requirements and other inspections by the FDA or other comparable regulatory authorities;
• our inability to negotiate manufacturing agreements with third parties under commercially reasonable terms, if at all;
21
• breach, termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us;
• reliance on single sources for drug components;
• lack of qualified backup suppliers for those components that are currently purchased from a sole or single-source supplier;
• our third-party manufacturers may not devote sufficient resources to our product candidates;
• we may not own, or may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing process for our product candidates;
• operations of our third-party manufacturers or suppliers could be disrupted by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier; and
• carrier disruptions or increased costs that are beyond our control.
In addition, if we enter into a strategic collaboration with a third party for the commercialization of our current or any future product candidates, we will not be able to control the amount of time or resources that they devote to such efforts. If any strategic collaborator does not commit adequate resources to the marketing and distribution of our product candidates, it could limit our potential revenues.
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize our current or any future product candidates once approved. Some of these events could be the basis for FDA action, including injunction, request for recall, seizure, or total or partial suspension of production.
We purchase components for our product candidates from third parties, some of which may be sole-source suppliers.
Our product candidate is comprised of three key ingredients, the excipient (referred to as SHAO) and two active, commercially available pharmaceutical ingredients cisplatin and vinblastine sulphate. Currently each of the three ingredients and our product candidate are single sourced. While we are aware of other suppliers for the two active ingredients, those suppliers have not been qualified as yet. We also have identified other producers of both the SHAO excipient and the product candidate. We manufacture SHAO using CuriaGlobal in Albany, New York and INT230-6 at CuriaGlobal in Glasgow, Scotland. We have only qualified CuriaGlobal to produce SHAO and INT230-6 at this time. We control the manufacturing processes for SHAO and INT230-6, and we have all information on the production of the molecule and product candidate; however, it would take several months to qualify a new supplier or suppliers. We purchase the cisplatin from Johnson Matthey in West Deptford, New Jersey. Johnson Matthey is the developer of cisplatin and one of the world’s largest producer of cisplatin. We have only qualified Johnson Matthey. We purchase vinblastine sulphate from Minakem located in Mont-Saint-Guibert, Belgium. We have only qualified Minakem as a supplier of our vinblastine sulphate. It would take several months to quality new vendors for cisplatin and vinblastine sulfate.
We rely and expect to continue to rely completely on third parties to manufacture key components of our preclinical, clinical trial and commercial product candidate supplies. The development and commercialization of any of our product candidates could be stopped, delayed or made less profitable if those third parties fail to provide us with sufficient quantities of such product supplies or fail to do so at acceptable quality levels, including in accordance with applicable regulatory requirements or contractual obligations, and our operations could be harmed as a result. The components of our product candidates, including enhancers, drugs, and excipients, must be manufactured and assembled in accordance with approved manufacturing and predetermined performance specifications and must meet CGMP and quality systems requirements. Some states also have similar regulations. Many of the other components of our product candidates may be manufactured by sole-source suppliers that may have proprietary manufacturing processes. If we need to find a new source of supply, we may face long interruptions in obtaining necessary components for our product candidates, in obtaining FDA or foreign regulatory agency approval of these components and in establishing the manufacturing process, which could jeopardize our ability to supply products using our technology to the market.
22
We have not entered into long term manufacturing and supply agreements with any producers.
We intend to pursue agreements with contract manufacturers to produce the components and drug products that we will use in the future for the commercialization of products that make using of our technology, as well as for labeling and finishing services. We may not be able to enter into such arrangements on acceptable terms or at all. Components of our product candidates are currently manufactured for us in small quantities for use in our preclinical and clinical studies. We will require significantly greater quantities to commercialize any given product. We may not be able to find alternate sources of comparable components. If we are unable to obtain adequate supplies of components from our existing suppliers or need to switch to an alternate supplier and obtain FDA or other regulatory agency approval of that supplier, commercialization of our product candidates may be delayed. If we are unable to obtain sufficient compounds and labeling services on acceptable terms, or if we should encounter delays or difficulties in our relationships with our current and future suppliers or if our current and future suppliers of each component do not comply with applicable regulations for the manufacturing and production of drugs, our business, financial condition, and results of operations may be materially harmed.
If we cannot successfully purchase or produce the drugs used in the manufacture of our product candidates, our ability to develop and commercialize products using our technology would be impaired.
To manufacture the therapeutic agents on our own, we would first have to develop a manufacturing facility that complies with FDA requirements and regulations to produce each therapeutic agent we choose to manufacture. Developing these resources would be an expensive and lengthy process and would have a material adverse effect on our revenues and profitability. We have no manufacturing history and we may not be able to scale up or demonstrate manufacture of commercial quantities, in a cost-effective manner, or in compliance with the regulatory requirements applicable to such manufacturing. Additionally, we may have difficulty obtaining other components for the system from our third-party suppliers in a timely manner or at all which may adversely affect our ability to conduct timely clinical trials in the United States and elsewhere to obtain regulatory approval, and our ability to deliver our product candidates to purchasers.
Our current and future relationships with investigators, health care professionals, consultants, third-party payors, and customers will be subject to applicable healthcare regulatory laws, which could expose us to penalties.
Our business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors, patient support, charitable organizations and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws regulate the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell, and distribute our product candidates for which we obtain marketing approval. Such laws include, among others: he federal Anti-Kickback Statute, the federal false claims laws, including the False Claims Act, the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, the federal Physician Payments Sunshine Act, federal consumer protection and unfair competition laws and analogous state and foreign laws and regulations, such as state antikickback and false claims laws, which may apply to our business practices. For additional information regarding the regulatory regime under which we operate, see “Business — Government Regulation.”
Efforts to ensure that our current and future business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable healthcare laws. If our operations are found to be in violation of any of these or any other health regulatory laws that may apply to us, we may be subject to significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs or similar programs in other countries or jurisdictions, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Even the mere issuance of a subpoena or the fact of an investigation alone, regardless of the merit, may result in negative publicity, a drop in our share price and other harm to our business, financial condition and results of operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
23
If we or any contract manufacturers and suppliers we engage fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our research and development activities involve the use of biological and hazardous materials and produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. We do not carry specific biological waste or hazardous waste insurance coverage, workers compensation or property and casualty and general liability insurance policies that include coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
We have limited experience in marketing and commercializing products and, as a result, we may not be successful in commercializing products made using our technology.
If we are unable to find a development or marketing partner, we may have to directly and indirectly market our product candidates. To pursue a direct marketing strategy in any country may require the engagement of a contract sales organization to provide medical science liaisons to educate the medical oncologists, and we may need to utilize a direct sales force to sell our product candidates to interventional radiologists and hospitals. However, we have not previously sold, marketed, or distributed any products and have limited experience in building a sales and marketing organization and in entering and managing relationships with third-party distributors. To pursue such a potential strategy, we must acquire or internally develop a sales, marketing, and distribution infrastructure and/or enter into strategic alliances to perform these services. The development of sales, marketing and distribution infrastructure is difficult and time consuming and would require substantial financial and other resources. If we cannot successfully partner the products for marketing or develop the infrastructure to market and commercialize the products ourselves, our ability to generate revenues may be harmed, and we may be required to enter strategic alliances to have such activities carried out on our behalf, which may not be on favorable terms.
Even if we are successful in commercializing products using our technology in the United States, we may not be successful in other foreign countries.
Each country requires a different commercialization strategy, so our U.S. strategy may not translate to other markets. Without a successful commercialization strategy tailored for each market, our efforts to promote and market the products in each of our target markets may fail in any or all those markets.
Our plan to use collaborative arrangements with third parties to help finance and to market and sell products using our technology may not be successful.
Our efforts may never result in the successful development or commercialization of products using our technology. The success of any development program will depend upon our ability to perform our obligations under any agreements as well as factors beyond our control, such as the commitment of our vendor collaborators and the timely
24
performance of their obligations. The terms of any such collaboration may permit our collaborators to abandon the alliance at any time for any reason or prevent us from terminating arrangements with vendors or collaborators who do not perform in accordance with our expectations or our collaborators may breach their agreements with us. In addition, any third parties with which we collaborate may have significant control over important aspects of the development and commercialization of our product candidates, including research and development, market identification, marketing methods, pricing, composition of sales force, and promotional activities. We are not able to control or influence the amount and timing of resources that any vendor or collaborator may devote to our research and development programs or the commercialization, marketing, or distribution of our product candidates. We may not be able to prevent any collaborators from pursuing alternative technologies or products that could result in the development of products that compete with our technology or the withdrawal of their support for our product candidates. The failure of any such collaboration could have a material adverse effect on our business.
We will be dependent on healthcare professionals’ efforts to learn about our product candidates.
As a result, the products being developed may not gain significant market acceptance among physicians, hospitals, patients and healthcare payors until healthcare professionals are properly educated about the procedures involved in using the products. Market acceptance of our product candidates and technology will depend upon a variety of factors including:
• whether our future clinical trials demonstrate significantly improved patient outcomes;
• our ability to educate and train physicians to perform the image guided injection procedures and drive acceptance of the use of products;
• our ability to convince healthcare payors that use of the technology results in reduced treatment costs and improved outcomes for patients;
• whether our system replaces and/or complements treatment methods in which many hospitals have made a significant investment; and
• whether doctors and hospitals are willing to replace their existing technology with a new medical technology until the new technology’s value has been demonstrated.
We may need to establish clinical training and centers of excellence to educate and train physicians and healthcare payors, but the key opinion thought leadership required for initial market acceptance within the healthcare arena may take time to develop.
Without effort from key opinion healthcare professionals to become educated about our product candidates, and guide physicians, the market may not accept our approach and our efforts to commercialize our product candidates may be unsuccessful. Similar considerations apply in any other market where we receive approval. Successful commercialization of the methodology in many markets will depend on market acceptance by thought leading healthcare professionals.
Healthcare legislative reform measures may have a negative impact on our business and results of operations.
In the United States and some foreign jurisdictions, there have been, and likely will continue to be, a number of legislative and regulatory changes and proposed changes regarding the healthcare system directed at broadening the availability of healthcare, improving the quality of healthcare, and containing or lowering the cost of healthcare. For example, in March 2010, the United States Congress enacted the ACA, which, among other things, includes changes to the coverage and payment for products under government health care programs. And since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the ACA.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2029 unless additional Congressional action is taken. The Coronavirus Aid, Relief, and Economic Security Act, or CARES Act, which was signed into law on March 27, 2020, designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended these reductions from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. In addition, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law,
25
which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single-source and innovator multiple-source drugs, beginning January 1, 2024. These laws may result in additional reductions in Medicare, Medicaid and other healthcare funding.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, such as bundled payment models. In addition, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products, which has resulted in several Congressional inquiries and proposed and enacted state and federal legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing and importation. As a result, the FDA also released a final rule in September 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, in November 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed by the Biden administration until January 1, 2023. The CMS also issued an interim final rule that establishes a Most Favored Nation, or MFN, Model for Medicare Part B drug payments. This regulation would substantially change the reimbursement landscape as it bases Medicare Part B payment for 50 selected drugs on prices in foreign countries instead of average sales prices (ASP) and establishes a fixed add-on payment in place of the current 6 percent (4.3 percent after sequestration) of ASP. The MFN drug payment amount is expected to be lower than the current ASP-based limit because U.S. drug prices are generally the highest in the world. On December 28, 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. On January 13, 2021, in a separate lawsuit brought by industry groups in the U.S. District Court for the District of Maryland, the government defendants entered a joint motion to stay litigation on the condition that the government would not appeal the preliminary injunction granted in the U.S. District Court for the Northern District of California and that performance for any final regulation stemming from the MFN Model interim final rule shall not commence earlier than sixty (60) days after publication of that regulation in the Federal Register. In December 2020, CMS issued a final rule implementing significant manufacturer price reporting changes under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to pharmacy benefit manager accumulator programs and Best Price reporting related to certain value-based purchasing arrangements. On May 21, 2021, an industry group sued CMS, claiming that the change to the Best Price rule exceeds CMS’s statutory authority and is contrary to the Medicaid Rebate statute. This litigation is ongoing. It is unclear to what extent these new regulations will be implemented and to what extent these regulations or any future legislation or regulations by the Biden administration will have on our business, including our ability to generate revenue and achieve profitability.
Outside the United States, ensuring coverage and adequate payment for a product also involves challenges. Pricing of prescription pharmaceuticals is subject to government control in many countries. Pricing negotiations with government authorities can extend well beyond the receipt of regulatory approval for a product and may require a clinical trial that compares the cost-effectiveness of a product to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in commercialization.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies or so-called health technology assessments, in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a product or they may instead adopt a system of direct or indirect controls on our profitability for placing the product on the market. Other member states allow companies to fix their own prices for products, but monitor and control prescription volumes and issue guidance to physicians to
26
limit prescriptions. Recently, many countries in the European Union have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union.
We expect that these and other healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug, which could have an adverse effect on demand for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates. For additional information on healthcare reform, see “Business — Government Regulation — Healthcare reform.”
Rapid technological developments in treatment methods for cancer and competition with other forms of cancer treatments could affect our ability to achieve meaningful revenues or profit.
Competition in the cancer treatment industry is intense. Products made using our technology will compete with all forms of cancer treatments that are alternatives to the “gold standard” treatment of surgical resection. Many of our competitors have substantially greater resources and considerable experience in conducting clinical trials and obtaining regulatory approvals. If these competitors develop more effective, more affordable products, or if treatment methods achieve earlier product development, our revenues or profitability will be substantially reduced.
The loss of key personnel could adversely affect our business.
The loss of any of our key members could delay our ability to develop the technology, conduct preclinical research, conduct clinical research, obtain FDA approval, or introduce products using our technology commercially and, ultimately, our ability to generate revenues and profits. Competition for experienced personnel is intense. If we cannot retain our current personnel or attract additional experienced personnel, our ability to compete could be adversely affected.
Risks Related to Patents, Trade Secrets, and Proprietary Rights
Our success depends in part on our ability to obtain patents, maintain trade secret protection, operate without infringing on the proprietary rights of third parties, and commercialize our technology prior to the expiration of our patent protection.
We have three U.S. patents and one pending U.S. patent application. We have 11 foreign patents, including one European patent, validated in 27 countries. We have five pending foreign patent applications. We have registered trademarks and know-how. While we have patents and filed patent applications covering composition of matter, use and methods, only 14 patents have issued. Due to the uncertainty of the patent prosecution process, there are no guarantees that our pending patent applications or any future applications will result in the issuance of a patent. Even if we are successful in obtaining more U.S. patents and new patents in other countries, there is no assurance that our patents will be upheld if later challenged or will provide significant protection or commercial advantage. Because of the length of time and expense associated with bringing new medical drugs and devices to the market, the healthcare industry has traditionally placed considerable emphasis on patent and trade secret protection for significant new technologies. Other parties may challenge our patents, patent claims or patent applications licensed or issued to us or may design around technologies we have patented, licensed or developed.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions such as patent term adjustments and/or extensions, may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including biosimilars. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
27
If we do not obtain patent term extension and data exclusivity for any product candidates we may develop, our business may be materially harmed.
Depending upon the timing, duration and specifics of any FDA marketing approval of any product candidates we may develop, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984 Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. However, we may not be granted an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our business, financial condition, results of operations, and prospects could be materially harmed.
Companies in the medical drug/device industry may use intellectual property infringement litigation to gain a competitive advantage.
In the United States, patent applications filed in recent years are confidential for 18 months, while older applications are not publicly available until the patent issues. As a result, even after the products using our technology are introduced to the market, there is no guarantee that we will be able to avoid patent infringement claims, whether such claims are ultimately held to have merit. Litigation may be necessary to enforce any patents issued or assigned to us or to determine the scope and validity of third-party proprietary rights. Litigation could be costly and could divert our attention from our business. There are no guarantees that we will receive a favorable outcome in any such litigation. If a third-party claims that we infringed its patents, any of the following may occur:
• we may become liable for substantial damages for past infringement if a court decides that our product candidates infringe upon a competitor’s patent;
• a court may prohibit us from selling or licensing our product candidates without a license from the patent holder, which may not be available on commercially acceptable terms or at all, or which may require us to pay substantial royalties or grant cross-licenses to our patents; and
• we may have to redesign our product candidate so that it does not infringe upon others’ patent rights, which may not be possible or could require substantial funds or time.
If a third party violates our intellectual property rights, we may be unable to enforce our rights because of our limited resources.
Use of our limited funds to enforce or to defend our intellectual property rights or to defend against legal proceedings alleging infringement of third party proprietary rights may also affect our financial condition adversely. If others file patent applications with respect to inventions for which we already have applications pending, we may be forced to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could also be costly and could divert our attention from our business. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before the any product can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent. Not all our U.S. patent rights will have corresponding patent rights effective in Europe or other foreign jurisdictions.
Similar considerations will apply in any other country where we may prosecute patent applications, may be issued patents, or may decide not to pursue patent protection relating to our technology. The laws of foreign countries may not protect our intellectual property rights to the same extent as do laws of the United States.
28
We protect our trade secrets and proprietary knowledge in part through confidentiality agreements with employees, consultants, and other parties. However, certain consultants, advisors and third parties with whom we have business relationships, and to whom in some cases we have disclosed or will disclose trade secrets and other proprietary knowledge, may also provide services to other parties in the medical device industry, including companies, universities, and research organizations that are developing competing products.
In addition, some employees may eventually seek employment with, and become employed by, our competitors. We cannot be assured that consultants, employees, and other third parties with whom we have entered into confidentiality agreements will not breach the terms of such agreements by improperly using or disclosing our trade secrets or other proprietary knowledge or that we will have adequate remedies for any such breach.
Trade secret protection does not prevent independent discovery of the technology or proprietary information or use of the same.
Competitors may independently duplicate or exceed our technology in whole or in part. If we are not successful in maintaining the confidentiality of our technology, the loss of trade secret protection or know-how relating to our technology will significantly impair our ability to commercialize our product candidates, and our value and results of operations will be harmed. Similar considerations apply in any other foreign country where we receive approval. Since we do not yet have valid issued patents for the products using our technology in some countries, our ability to successfully commercialize our technology in those countries may be harmed.
Risks Related to Products Liability
We may be the subject of product liability claims or product recalls, and we may be unable to maintain insurance adequate to cover potential liabilities.
Our business exposes us or may in the future expose us to potential liability risks that may arise from the testing, manufacture, marketing, sale and use of products using our technology. In addition, because certain products using the new technology are intended for use in patients with cancer, there is an increased risk of death among the patients treated with our system which may increase the risk of product liability lawsuits. We may be subject to claims against us even if the injury is due to the actions of others. For example, if the medical personnel that use our product candidates on patients are not properly trained or are negligent in the use of our product candidates, the patient may be injured through the use of our product candidates, which may subject us to claims. Were such a claim asserted we would likely incur substantial legal and related expenses even if we prevail on the merits. Claims for damages, whether or not successful, could cause delays in clinical trials and result in the loss of physician endorsement, adverse publicity and/or limit our ability to market and sell the system, resulting in loss of revenue. In addition, it may be necessary for us to recall products that do not meet approved specifications, which would also result in adverse publicity, as well as resulting in costs connected to the recall and loss of revenue. A successful products liability claim, or product recall would have a material adverse effect on our business, financial condition and results of operations. We currently carry product liability and clinical trial insurance coverage, but it may be insufficient to cover one or more large claims.
Risks Related to Our Common Stock and This Offering
We are an “emerging growth company” as defined in the JOBS Act and a “smaller reporting company” as defined in the Securities Exchange Act of 1934, as amended, or the Exchange Act, and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies, which could make our common stock less attractive to investors and adversely affect the market price of our common stock.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of the fiscal year following the fifth anniversary of the date of the completion of this offering; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the Securities and Exchange Commission, which means the market value of our common stock
29
that is held by non-affiliates exceeds $700 million as of the prior June 30. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
• not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404;
• not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements;
• providing only two years of audited financial statements in addition to any required unaudited interim financial statements and a correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure;
• reduced disclosure obligations regarding executive compensation; and
• exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In this prospectus, we have not included all of the executive compensation-related information that would be required if we were not an emerging growth company.
We may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of reduced reporting burdens in this prospectus. In particular, we have provided only two years of audited financial statements and have not included all of the executive compensation information that would be required if we were not an emerging growth company. We cannot predict whether investors will find our common stock less attractive if we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to use the extended transition period for new or revised accounting standards during the period in which we remain an emerging growth company; however, we may adopt certain new or revised accounting standards early.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we no longer qualify as an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies until the fiscal year following the determination that our voting and non-voting common stock held by non-affiliates is more than $250 million measured on the last business day of our second fiscal quarter, or our annual revenues are more than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is more than $700 million measured on the last business day of our second fiscal quarter.
So long as we qualify as an “emerging growth company” or a “smaller reporting company,” we may elect not to provide you with certain information, including certain financial information and certain information regarding compensation of our executive officers, that we would otherwise have been required to provide in filings we make with the SEC, which may make it more difficult for investors and securities analysts to evaluate our company. Further, as mentioned above, so long as we qualify as an “emerging growth company” our independent registered public accounting firm will not be required to provide an attestation report on the effectiveness of our internal control over financial reporting, which may increase the risk that material weaknesses or significant deficiencies in our internal control over financial reporting go undetected. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile and may decline.
30
Substantial influence will remain with our management and major stockholder, which could delay or prevent a change of control or cause us to take actions in conflict with the intent of our stockholders.
Immediately following the completion of this offering, and disregarding any shares of common stock that they purchase in this offering, the existing holdings of our executive officers, directors, principal stockholders and their affiliates will represent beneficial ownership, in the aggregate, of approximately 61.6% of our outstanding common stock. We anticipate that our President and CEO will be our largest overall shareholder following the completion of this offering, beneficially owning approximately 23.4% of our outstanding common stock. These stockholders, if they act together, will be able to influence our management and affairs and the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. These stockholders acquired their shares of common stock for substantially less than the price of the shares of common stock being acquired in this offering, and these stockholders may have interests with respect to their common stock that are different from those of investors in this offering. The concentration of voting power among these stockholders may have an adverse effect on the price of our common stock.
An active trading market for our common stock may not develop, and you may not be able to resell your shares at or above the initial public offering price.
Prior to this offering, there has been no public market for shares of our common stock. Although we anticipate that our common stock will be approved for listing on The Nasdaq Capital Market, or Nasdaq, an active trading market for our shares may never develop or be sustained following this offering. The initial public offering price of our common stock will be determined through negotiations between us and the underwriters. This initial public offering price may not be indicative of the market price of our common stock after this offering. In the absence of an active trading market for our common stock, investors may not be able to sell their common stock at or above the initial public offering price or at the time that they would like to sell.
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock in this offering.
Our stock price is likely to be volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility. Due to our history of losses as well as a variety of factors, many of which are outside of our control and may be difficult to predict, our quarterly and annual operating results may fluctuate significantly in the future. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially.
Further, investors in our common stock may experience a decrease, which could be substantial, in the value of their stock for reasons unrelated to our operating performance or prospects, and could lose part or all of their investment. The price of our common stock could be subject to wide fluctuations in response to a number of factors, including those described elsewhere in this prospectus and others such as:
• variations in our operating performance and the performance of our competitors;
• actual or anticipated fluctuations in our quarterly or annual operating results;
• publication of research reports by securities analysts about us or our competitors or our industry;
• announcements by us, our competitors or our vendors of significant contracts, acquisitions, joint marketing relationships, joint ventures or capital commitments;
• our failure or the failure of our competitors to meet analysts’ projections or guidance that we or our competitors may give to the market;
• additions and departures of key personnel;
• strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in business strategy;
31
• the passage of legislation or other regulatory developments affecting us or our industry;
• speculation in the press or investment community;
• changes in accounting principles;
• terrorist acts, acts of war or periods of widespread civil unrest;
• natural disasters and other calamities; and
• changes in general market and economic conditions.
As a result of this volatility, you may not be able to sell your common stock at or above the initial public offering price.
If you purchase our common stock in this offering, you will incur immediate and substantial dilution in the book value of your shares.
You will suffer immediate and substantial dilution in the net tangible book value of the common stock you purchase in this offering. Based on the initial public offering price of $7.00 per share, purchasers of common stock in this offering will experience immediate dilution of $5.95 per share in net tangible book value of the common stock. In addition, investors purchasing common stock in this offering will contribute 32.1% of the total amount invested by stockholders since inception but will only own 12.2% of the shares of common stock outstanding. In the past, we issued options and other securities to acquire common stock at prices significantly below the initial public offering price. To the extent these outstanding securities are ultimately exercised, investors purchasing common stock in this offering will sustain further dilution. See “Dilution” for a more detailed description of the dilution to new investors in the offering.
Sales of a substantial number of shares of our common stock by our existing stockholders in the public market could cause our stock price to fall.
Upon the closing of this offering, we will have outstanding a total of 17,594,053 shares of common stock (or 17,915,481 shares if the underwriters exercise in full their option to purchase additional shares of common stock). All the common stock sold in this offering will be freely transferable, except for any shares held by our “affiliates,” as that term is defined in Rule 144 under the Securities Act of 1933, as amended (the “Securities Act”). In addition, each of our directors and executive officers and four of our equity holders have entered into a lock-up agreement with A.G.P./Alliance Global Partners, as representative on behalf of the underwriters, which regulates their sales of our common stock for a period of 180 days after the date of this prospectus, subject to certain exceptions and automatic extensions in certain circumstances. See the section entitled “Shares Eligible for Future Sale — Lock-Up Agreements” in this prospectus.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after the lockup and other legal restrictions on resale discussed in this prospectus lapse, the trading price of our common stock could decline.
Our management will have broad discretion in using the proceeds of this offering and may not use these proceeds effectively, which could affect our results of operations and cause our stock price to decline.
We will have considerable discretion in the application of the net proceeds of this offering. We intend to use the net proceeds from this offering to fund discovery and clinical development efforts as well as to further expand our manufacturing platform and capabilities, to grow our infrastructure to support our pipeline, and to fund new and ongoing research activities, working capital and other general corporate purposes, which may include funding for the hiring of additional personnel, capital expenditures and the costs of operating as a public company. As a result, investors will be relying upon management’s judgment with only limited information about our specific intentions for the use of the balance of the net proceeds of this offering. We may use the net proceeds for purposes that do not yield a significant return or any return at all for our stockholders. In addition, pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses value.
32
We do not anticipate paying dividends in the foreseeable future.
We do not anticipate paying dividends on our common stock in the foreseeable future. Therefore, in the absence of an acquisition transaction, the only way to realize a return on investment might be for investors to sell the stock, but it is unknown when, if ever, investors will be able to do so.
Provisions in our charter documents and Delaware law may deter takeover efforts that could be beneficial to stockholder value.
Our certificate of incorporation and by-laws and Delaware law contain provisions that could make it harder for a third party to acquire us, even if doing so might be beneficial to our stockholders. These provisions include a classified board of directors and limitations on actions by our stockholders. In addition, our board of directors has the right to issue preferred stock without stockholder approval that could be used to dilute a potential hostile acquiror. Our certificate of incorporation also imposes some restrictions on mergers and other business combinations between us and any holder of 15.0% or more of our outstanding common stock. As a result, you may lose your ability to sell your stock for a price in excess of the prevailing market price due to these protective measures, and efforts by stockholders to change our direction or management may be unsuccessful. See the section entitled “Description of Capital Stock” in this prospectus.
Our amended and restated certificate of incorporation that will be in effect at the closing of this offering will provide that the Court of Chancery of the State of Delaware will be the exclusive forum for certain disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation that will be in effect immediately prior to the closing of this offering will provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware is the exclusive forum for the following types of actions or proceedings under Delaware statutory or common law:
• any derivative claim or cause of action brought on our behalf;
• any claim or cause of action for a breach of fiduciary duty owed by any of our current or former directors, officers or other employees to us or our stockholders;
• any claim or cause of action against us or any of our current or former directors, officers or other employees arising out of or pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation or our bylaws (as each may be amended from time to time);
• any claim or cause of action seeking to interpret, apply, enforce or determine the validity of our amended and restated certificate of incorporation or our amended and restated bylaws (as each may be amended from time to time, including any right, obligation or remedy thereunder);
• any claim or cause of action as to which the Delaware General Corporation Law confers jurisdiction to the Court of Chancery of the State of Delaware; and
• any claim or cause of action against us or any of our current or former directors, officers or other employees governed by the internal-affairs doctrine.
This provision would not apply to suits brought to enforce a duty or liability created by the Securities Act, the Exchange Act or any other claim for which the U.S. federal courts have exclusive jurisdiction. In addition, unless we consent in writing to the selection of an alternative forum, to the fullest extent permitted by law, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause or causes of action arising under the Securities Act, including all causes of action asserted against any defendant to such complaint.
For the avoidance of doubt, this provision is intended to benefit and may be enforced by us, our officers and directors, the underwriters to any offering giving rise to such complaint, and any other professional entity whose profession gives authority to a statement made by that person or entity and who has prepared or certified any part of the documents underlying the offering. However, these choice of forum provisions may limit a stockholder’s ability
33
to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, or other employees. Further, these choice of forum provisions may increase the costs for a stockholder to bring such a claim and may discourage them from doing so.
While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions, and there can be no assurance that such provisions will be enforced by a court in those other jurisdictions. If a court were to find the choice of forum provision contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions. For example, the Court of Chancery of the State of Delaware recently determined that the exclusive forum provisions of federal district courts of the United States of America for resolving any complaint asserting a cause of action arising under the Securities Act is not enforceable. We note that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder.
Our board of directors could issue additional shares of common stock or a new class of preferred stock and dilute the equity positions of current stockholders without consent of the investors.
We are issuing new shares of preferred stock as part of this offering. In the future, we expect to need additional funding, which it may obtain through the authorization and issuance of additional common or preferred equity securities. The authorization of additional shares of stock under our certificate of incorporation may be made without the affirmative vote of all the investors in this offering. Any issuance of additional shares of stock could dilute the equity position of our current stockholders. A future issuance of shares of preferred stock will result in the shares of our common stock being subject to certain preferential rights of such preferred stock, including a right to participate in the proceeds of any sale or liquidation of the Company ahead of the shares of common stock.
THE SELECTED LIST OF RISK FACTORS ABOVE DOES NOT PURPORT TO BE A COMPLETE LIST OF ALL MATERIAL RISKS INHERENT WITH AN INVESTMENT IN THIS OFFERING. WE URGE YOU TO CAREFULLY CONSIDER THESE RISKS AS WELL AS OTHERS COMMON TO EARLY STAGE VENTURES AND OTHER INVESTMENTS OF SIMILAR NATURE.
34
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, contained in this prospectus, including statements regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “will,” “project,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These forward-looking statements include, among other things, statements about:
• the initiation, timing, progress and results of future preclinical studies and clinical trials, and our research and development programs;
• our need to raise additional funding before we can expect to generate any revenues from product sales;
• our plans to develop and commercialize our product candidates;
• the timing or likelihood of regulatory filings and approvals;
• the ability of our research to generate and advance additional product candidates;
• the implementation of our business model, strategic plans for our business, product candidates and technology;
• our commercialization, marketing and manufacturing capabilities and strategy;
• the rate and degree of market acceptance and clinical utility of our system;
• our competitive position;
• our intellectual property position;
• developments and projections relating to our competitors and our industry;
• our ability to maintain and establish collaborations or obtain additional funding;
• our expectations related to the use of proceeds from this offering; and
• our estimates regarding expenses, future revenue, capital requirements and needs for additional financing.
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in this prospectus, particularly in the “Risk Factors” section, that could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments that we may make. You should read this prospectus completely and with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this prospectus and the documents that we have filed with the SEC as exhibits to the registration statement, of which this prospectus is a part, with the understanding that our actual future results, levels of activity, performance and events and circumstances may be materially different from what we expect. We qualify all forward-looking statements by these cautionary statements.
35
We estimate that the net proceeds from the sale of our common stock in this offering will be approximately $13.1 million, based upon an assumed initial public offering price of $10.00 per share, the midpoint of the price range set forth on the cover page of this prospectus, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters’ option to purchase additional shares of our common stock is exercised in full, we estimate that the net proceeds from the offering will be approximately $15.1 million.
We intend to use the net proceeds from this offering for the following purposes:
• approximately 50% toward conducting the Phase 3 sarcoma study (IT-03);
• approximately 25% toward our current clinical trials and related operations, including costs associated with the manufacturing of SHAO and INT230-6 for our clinical trials, conducting our existing clinical programs, initiating a metastatic breast cancer phase 2 study, and maintaining our IND with the FDA, CTA with Health Canada as well as other regulatory filings with other countries;
• approximately 5% toward development of our second product candidate, INT33X; and
• approximately 20% toward general corporate purposes and working capital.
Based on our current plans, we believe that our existing cash and cash equivalents, together with the anticipated net proceeds to us from this offering, will enable us to fund our operations and capital expenditure requirements through September 2023.
We will have broad discretion over how to use the net proceeds we receive from this offering. The expected uses of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our development, the status of and results from preclinical and clinical trials, and any unforeseen cash needs.
36
We have not declared or paid any cash dividends on our capital stock since our inception. In the near term we intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends to holders of common stock in the foreseeable future.
37
The following table sets forth our cash and cash equivalents and consolidated capitalization as of September 30, 2021:
• on an actual basis;
• on a pro forma basis to give effect to:
• the automatic conversion of 8,249,719 outstanding shares of our preferred stock into an aggregate of 8,249,719 shares of our common stock upon the closing of this offering;
• the conversion of a convertible note into an aggregate of 381,265 shares of our common stock upon the closing of this offering, which is based on unpaid principal and accrued but unpaid interest as of September 30, 2021 at a conversion price of $5.25 per share; and
• the filing and effectiveness of our amended and restated certificate of incorporation, which will occur immediately prior to the consummation of this offering; and
• and on a pro forma as adjusted basis, giving effect to our issuance and sale of shares of common stock in this offering at an assumed initial public offering price of $7.00 per share (the midpoint of the price range set forth on the cover page of this prospectus), after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us, and the application of the proceeds as described under the section entitled “Use of Proceeds”.
This table should be read in conjunction with the other information contained in this prospectus, including “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and related notes appearing elsewhere in this prospectus.
|
At September 30, 2021 |
||||||||||||
|
ACTUAL |
PRO FORMA |
PRO FORMA AS ADJUSTED |
||||||||||
|
(In thousands, except share and per share data) |
||||||||||||
|
Cash and cash equivalents |
$ |
7,408 |
|
$ |
7,408 |
|
$ |
20,458 |
|
|||
|
Series A redeemable convertible preferred stock, par value $0.0001, 5,000,000 shares authorized, issued and outstanding, actual; no shares issued or outstanding, pro forma and pro forma as adjusted |
|
10,000 |
|
|
— |
|
|
— |
|
|||
|
Stockholders’ equity (Deficit) |
|
|
|
|
|
|
||||||
|
Series B convertible preferred stock, par value $0.0001, 1,449,113 shares authorized, issued and outstanding, actual; no shares issued or outstanding, pro forma and pro forma as adjusted |
|
— |
|
|
— |
|
|
— |
|
|||
|
Series C convertible preferred stock, par value $0.0001, 1,800,606 shares authorized, issued and outstanding, actual; no shares issued or outstanding, pro forma and pro forma as adjusted. |
|
— |
|
|
— |
|
|
— |
|
|||
|
Common stock, par value $.0001, 50,000,000 shares authorized, 6,820,211 shares issued and outstanding, actual; 135,000,000 shares authorized, 15,451,195 shares issued and outstanding, pro forma; 135,000,000 shares authorized, 17,594,053 shares issued and outstanding, pro forma as adjusted. |
|
1 |
|
|
1 |
|
|
2 |
|
|||
|
Additional paid-in capital |
|
22,130 |
|
|
34,131 |
|
|
47,180 |
|
|||
|
Accumulated deficit |
|
(28,670 |
) |
|
(28,670 |
) |
|
(28,670 |
) |
|||
|
|
|
|
|
|
|
|||||||
|
Total stockholders’ equity (deficit) |
|
(6,539 |
) |
|
5,462 |
|
|
18,512 |
|
|||
|
|
|
|
|
|
|
|||||||
|
Total capitalization |
$ |
3,461 |
|
$ |
5,462 |
|
$ |
18,512 |
|
|||
38
The information set forth in the table excludes:
• 1,822,500 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2021 under our 2013 Stock and Option Plan, or the 2013 Plan, at a weighted average exercise price of $4.28 per share; of these, 1,152,250 were exercisable at a weighted average exercise price of $3.52 per share;
• 2,677,500 shares of our common stock reserved and available for future issuance under the 2013 Plan, as of September 30, 2021, which will cease to be available for issuance at the time that our 2021 Stock Incentive Plan, or the 2021 Plan, becomes effective;
• 646,500 shares of our common stock reserved and available for future issuance upon exercise of the outstanding warrants, as of September 30, 2021 at a weighted average exercise price of $3.00 per share; of these 568,974 were exercisable at a weighted average exercise price of $2.68 per share; and
• 3,000,000 shares of our common stock that will become available for future issuance under the 2021 Plan, which will become effective in connection with the completion of this offering.
Unless otherwise indicated, all information contained in this prospectus assumes no exercise by the underwriters of their option to purchase additional shares and no exercise of any other options or warrants.
39
If you invest in our common stock in this offering, your ownership interest will be diluted immediately to the extent of the difference between the initial public offering price per share of our common stock and the pro forma as adjusted net tangible book value per share of our common stock after this offering.
Our historical net tangible book value (deficit) as of September 30, 2021 was ($6.5 million), or ($0.96) per share of our common stock. Our historical net tangible book value (deficit) is the amount of our total tangible assets less our total liabilities and preferred stock, which is not included within our stockholders’ (deficit) equity. Historical net tangible book value per share represents historical net tangible book value (deficit) divided by the 6,820,211 shares of our common stock outstanding as of September 30, 2021. This calculation of historical net tangible book value (deficit) includes $2.0 million in convertible notes and $10.0 million in Series A preferred stock, both of which convert into common stock at an IPO of at least $15.0 million.
The pro forma data in the table below is derived from our balance sheet as of September 30, 2021 and is presented on a pro forma basis after giving effect to the conversion of our preferred stock into 8,249,719 shares of our common stock and the issuance of 381,265 shares of our common stock upon the conversion of a convertible note and accrued interest as of September 30, 2021 at a conversion price of $5.25 per share, each of which is expected to occur at the closing of this offering. Our pro forma net tangible book value (deficit) as of September 30, 2021 was $5.5 million, or $0.36 per share of our common stock. Our pro forma net tangible book value (deficit) is the amount of our total tangible assets less our total liabilities. Pro forma net tangible book value per share represents pro forma net tangible book value (deficit) divided by the 15,451,195 shares of our common stock outstanding as of September 30, 2021 after giving effect to these pro forma adjustments.
After giving effect to the receipt of the estimated net proceeds from our sale of common stock in this offering, assuming an initial public offering price of $7.00 per share (the mid-point of the offering range shown on the cover of this prospectus), after deducting the underwriting discount and other estimated offering expenses payable by us, our pro forma and as-adjusted net tangible book value as of September 30, 2021 would have been approximately $18.5 million, or $1.05 per share. This represents an immediate increase in pro forma as adjusted net tangible book value per share of $0.69 to existing stockholders and immediate dilution to new investors in this offering of $5.95 per share. The following table illustrates this dilution per share. Dilution represents the difference between the amount per share paid by investors in this offering and the pro forma and as-adjusted net tangible book value per share of our common stock immediately after this offering.
|
Assumed initial public offering price per share |
|
|
$ |
7.00 |
|||
|
Historical net tangible book value (deficit) per share as of September 30, 2021 |
$ |
(0.96 |
) |
|
|||
|
Increase per share attributable to the pro forma adjustment described above |
|
1.32 |
|
|
|||
|
Pro forma net tangible book value per share as of September 30, 2021 |
|
0.36 |
|
|
|||
|
Increase in net tangible book value per share attributable to new investors in this offering |
|
0.69 |
|
|
|||
|
Pro forma and as-adjusted net tangible book value per share after this offering |
|
|
|
1.05 |
|||
|
Dilution per share to new investors |
|
|
$ |
5.95 |
If the underwriters fully exercise their option to purchase additional shares, pro forma and as-adjusted net tangible book value after this offering would increase by approximately $0.11 per share, and there would be an immediate dilution of approximately $5.84 per share to new investors based on the initial public offering price of $7.00 per share, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. If any shares are issued upon exercise of outstanding options or warrants, you will experience further dilution.
The dilution information discussed above is illustrative only and will change based on the actual initial public offering price and other terms of this offering determined at pricing. A $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share of our common stock, the midpoint of the price range set forth on the cover page of this prospectus, would decrease (increase) our pro forma and as-adjusted net tangible book value after giving effect to the offering by $1.993 million, assuming no change to the number of shares of our common stock offered by us as set forth on the cover page of this prospectus, and after deducting estimated underwriting discounts and commissions and estimated expenses payable by us.
40
Each increase of 200,000 shares in the number of shares offered by us would increase our pro forma and as-adjusted net tangible book value by $1.30 million, increase the pro forma and as-adjusted net tangible book value per share after this offering by $0.07 and decrease the dilution per share to new investors by $0.07, assuming the assumed public offering price remains the same and after deducting the estimated underwriting discounts and commissions and estimated expenses payable by us. Each decrease of 200,000 shares in the number of shares offered by us would decrease our pro forma and as-adjusted net tangible book value by $1.30 million, decrease the pro forma and as-adjusted net tangible book value per share after this offering by $0.07 and increase the dilution per share to new investors by $0.07, assuming the assumed public offering price remains the same and after deducting the estimated underwriting discounts and commissions and estimated expenses payable by us.
The following table presents, on a pro forma as-adjusted basis, as described above, the differences between the existing stockholders on a pro forma basis and the purchasers of shares of common stock in this offering with respect to the number of shares of common stock purchased from us, the total consideration paid, and the average price paid per share at an assumed public offering price of $7.00 per share (the midpoint of the range set forth on the cover page of this prospectus):
|
Shares Purchased |
Total Consideration |
Average |
||||||||||
|
Number |
Percent |
Amount |
Percent |
|||||||||
|
Existing stockholders |
15,451,195 |
87.8 |
$ |
31,750,850 |
67.9 |
$ |
2.05 |
|||||
|
New investors |
2,142,858 |
12.2 |
|
15,000,006 |
32.1 |
|
7.00 |
|||||
|
Total |
17,594,053 |
100.0 |
|
46,750,856 |
100.0 |
|
2.66 |
|||||
The table above assumes no exercise of the underwriters’ option to purchase additional shares in this offering. If the underwriters’ option to purchase additional shares is exercised in full, the number of shares of our common stock held by existing stockholders would be reduced to 86.2% of the total number of shares of our common stock outstanding after this offering, and the number of shares of common stock held by new investors participating in this offering would be increased to 13.8% of the total number of shares of our common stock outstanding after this offering.
The number of shares of our common stock to be outstanding after this offering is based on 15,451,195 shares of common stock outstanding as of September 30, 2021, which includes 6,820,211 shares of our common stock outstanding as of September 30, 2021, plus 8,249,719 shares of our common stock issued upon the conversion of our preferred stock and 381,265 shares of our common stock that would be issued on the convertible note and accrued interest as of September 30, 2021 at a conversion price of $5.25 per share and excludes:
• 1,822,500 shares of our common stock issuable upon the exercise of stock options outstanding as of September 30, 2021 under the 2013 Plan at a weighted average exercise price of $4.28 per share. Of these, 1,152,250 are exercisable at September 30, 2021 at a weighted average exercise price of $3.52 per share;
• 2,677,500 shares of our common stock reserved and available for future issuance under the 2013 Plan, as of September 30, 2021, which will cease to be available for issuance at the time that the 2021 Plan becomes effective;
• 646,500 shares of our common stock reserved and available for future issuance upon exercise of the outstanding warrants, as of September 30, 2021 at a weighted average exercise price of $3.00 per share. Of these, 568,974 are exercisable at September 30, 2021 at a weighted average exercise price of $2.68 per share; and
• 3,000,000 shares of our common stock that will become available for future issuance under the 2021 Plan, which will become effective in connection with the completion of this offering.
Unless otherwise indicated, all information contained in this prospectus assumes no exercise by the underwriters of their option to purchase additional shares and no exercise of any other options or warrants.
To the extent that outstanding options are exercised or shares are issued under our 2021 Plan, you will experience further dilution. In addition, we may choose to raise additional capital due to market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of these securities may result in further dilution to our stockholders.
41
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read together with our financial statements and related notes and other financial information appearing elsewhere in this prospectus. This discussion and analysis contains forward-looking statements that involve risk, uncertainties and assumptions. See the section entitled “Cautionary Note Regarding Forward-Looking Statements” in this prospectus. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of many factors, including those discussed in “Risk Factors” and elsewhere in this prospectus.
Unless otherwise indicated or the context otherwise requires, references in this prospectus to the “Company”, “we”, “us” and “our” refer to Intensity Therapeutics, Inc.
Overview
We are a clinical-stage biotechnology company whose treatment approach addresses both the regional and systemic nature of a patient’s cancer. We are currently in Phase 2 clinical trials with one study using our lead product candidate in late stage disease and another study in early stage breast cancer. Our first clinical trial has dosed 95 patients through September 30, 2021. This clinical trial is monotherapy using our lead product candidate INT230-6; a combination study with Merck’s Keytruda (pembrolizumab) for patients with advanced solid malignancies including pancreatic, bile duct, squamous cell, and non-MSI high colon cancers; and a combination study with Bristol Myers Squibb’s Yervoy (ipilimumab) for patients with breast cancer, liver cancer, and advanced sarcoma. Our second clinical trial has dosed 20 patients. This clinical trial is a Phase 2 randomized, window of opportunity study in early stage breast cancer. The primary endpoint is the proportion of patients who achieve a complete cell cycle arrest, defined as a reduction in the proportion of cells staining positive for Ki67, a widely used marker of cancer cell proliferation, as assessed by immunohistochemistry.
Since our inception in 2012, our operations have included business planning, hiring personnel, raising capital, building our intellectual property portfolio, and performing both research and development on our product candidates. We currently have incurred net losses since inception and expect to incur net losses in the future as we continue our research and development activities. To date, we have funded out operations primarily through approximately $32.1 million of sales of our common, preferred stock and convertible notes. As of September 30, 2021, we have approximately $7.4 million of cash and cash equivalents. Since our inception we have incurred significant operating losses. We incurred net losses of $5.5 million and $4.4 million for the nine months ended September 30, 2021 and 2020, respectively. As of September 30, 2021 and 2020, we had an accumulated deficit of approximately $28.7 million and $21.6 million, respectively. We expect to incur significant expenses and operating losses for the next several years. See “Funding Requirements” below.
We expect our expenses to increase as we continue to:
• Initiate Phase 3 programs in sarcoma and breast cancer;
• Continue to enroll patients into our current Phase 2 programs;
• Advance our preclinical research and bring new product into clinical development;
• Incur manufacturing costs for additional GMP batches of our product candidates and enhancer molecules;
• Seek regulatory approvals for any of our product candidates that successfully complete clinical trials;
• Hire additional personnel;
• Expand our operational, financial, and management systems;
• Invest in measures to protect our existing and new intellectual property;
• Establish a sales, marketing, medical affairs, and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval and intend to commercialize; and
• Operate as a public company.
42
Due to numerous risks and uncertainties associated with biopharmaceutical product development and the economic and developmental uncertainty arising from the COVID-19 pandemic, we may be unable to accurately predict the timing or magnitude of all expenses. Our ability to ultimately generate revenue to achieve profitability will depend heavily on the development, approval, and subsequent commercialization of our product candidates. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
As a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through the sale of equity, debt financing, or other capital sources, which may include collaborations with other companies or other strategic transactions. We may not be able to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as and when needed, we would have to significantly delay, reduce, or eliminate the development and commercialization of one or more of our product candidates.
Components of Results of Operations
Revenue
To date, we have not generated any revenue from product sales and we do not expect any revenue from the sale of product in the foreseeable future. If our development efforts for any of our product candidates are successful and result in regulatory approval, then we may generate revenue in the future from product sales. We cannot predict if, when, or to what extent we will generate revenue from the commercialization and sale of any of our product candidates. We may never succeed in obtaining regulatory approval for any of our product candidates.
Research and Development Costs
Salaries and Payroll Taxes
Salaries and payroll taxes include Company employees involved in our pre-clinical research and clinical trials. This includes medical officers, project management, manufacturing staff and research scientists. The payroll taxes include all government required payments such as social security and unemployment taxes.
Fringe Benefits
We offer a partially funded health insurance and dental insurance plan. We maintain a defined contribution plan for all employees age 21 and older who have completed one year of service. This 401K plan makes a matching contribution equal to 100% of an employee’s contribution, up to 3% of an employee’s eligible earnings.
Research Costs
Research costs include:
• Pre-clinical research
• Manufacture of new enhancer compounds,
• Manufacture and labeling of GMP product candidate
• Product candidate stability testing of GMP batches
• Costs due to clinical trial sites for patient care
• Other clinical trial costs such as shipping, storage, and analytical testing
Scientific Consulting
Scientific consulting are costs related to non-employees involved in research. This includes: statistical analysis, development of product manufacturing techniques, and internet research related to oncology and chemistry issues that may impact our preclinical or clinical research.
43
Stock-Based Compensation
Stock-based compensation is the expense related to stock options granted to our employees and warrants granted to our independent consultants who work in the research aspects.
General and Administrative Costs
Salaries and Payroll Taxes
Salaries and payroll taxes includes Company employees who are involved in fund raising, management, and our financial administration. The payroll taxes include all government required payments such as social security and unemployment taxes.
Fringe Benefits
We offer a partially funded health insurance and dental insurance plan. We maintain a defined contribution plan for all employees age 21 and older who have completed one year of service. This 401K plan makes a matching contribution equal to 100% of an employee’s contribution, up to 3% of an employee’s eligible earnings.
Legal
Legal costs relate primarily to our corporate administration. All legal costs relate to expenses for our outside corporate law firm.
Patent and Trademark
Patent and Trademark are the legal costs and filing costs to establish and maintain patents in 37 countries.
Recruiting
Recruiting is costs paid to outside consultants to identify and hire qualified new employees. Recruiting costs also include the cost of conducting background checks on potential employees.
Insurance
Insurance includes: directors and officers insurance, workers compensation insurance, product liability insurance, business insurance, employee and cyber liability insurance.
Facilities and Rent
Facilities and rent is the cost of maintaining our office facility in Westport, Connecticut.
Investor Relations
Investor relations are costs paid to outside consultants to develop the materials to present to prospective investors, and to arrange meetings with potential investors.
Public Relations
Public relations are costs related to press releases, obtaining mainstream and social media coverage, and our other public awareness.
Accounting Services
Accounting services include the cost of our independent auditors, costs related to the preparation of income tax returns, and the cost of maintain our accounting system.
Other
Other general and administrative costs include such items as office supplies, computer related costs, bank fees, and conferences.
44
Stock-Based Compensation
Stock-based compensation is the expense related to stock options granted to our employees and warrants granted to our independent consultants who work in the general and administrative aspects.
Other income
We earned interest income on its excess cash. All investments are in U.S. Treasury bills and bank certificates of deposit.
We received a refundable Connecticut Research and Development tax credit and a Federal Research and Development tax credit that is recoverable through a refund of Social Security taxes paid by us.
Results of Operations
The following table summarizes our results of operations for the years ended December 31, 2019 and 2020 and the nine months ended September 30, 2020 and 2021 (in thousands):
|
YEAR ENDED DECEMBER 31, |
INCREASE/ |
NINE MONTHS ENDED SEPTEMBER 30, |
INCREASE/ |
|||||||||||||||||||||
|
2020 |
2019 |
2021 |
2020 |
|||||||||||||||||||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
Research and development |
$ |
5,050 |
|
$ |
4,437 |
|
$ |
613 |
|
$ |
4,419 |
|
$ |
3,644 |
|
$ |
775 |
|
||||||
|
General and administrative |
|
1,173 |
|
|
1,238 |
|
|
(65 |
) |
|
1,184 |
|
|
922 |
|
|
262 |
|
||||||
|
Total operating expenses |
|
6,223 |
|
|
5,675 |
|
|
548 |
|
|
5,603 |
|
|
4,566 |
|
|
1,037 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
Loss from operations |
|
(6,223 |
) |
|
(5,675 |
) |
|
(548 |
) |
|
(5,603 |
) |
|
(4,566 |
) |
|
(1,037 |
) |
||||||
|
Other income |
|
193 |
|
|
295 |
|
|
(102 |
) |
|
109 |
|
|
170 |
|
|
(61 |
) |
||||||
|
Net loss |
$ |
(6,030 |
) |
$ |
(5,380 |
) |
$ |
(650 |
) |
$ |
(5,494 |
) |
$ |
(4,396 |
) |
$ |
(1,098 |
) |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
Loss per share, basic and diluted |
$ |
(0.88 |
) |
$ |
(0.79 |
) |
|
|
$ |
(0.81 |
) |
$ |
(0.64 |
) |
|
|
||||||||
|
Weighted average number of shares |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
Stock, basic and diluted |
|
6,819,026 |
|
|
6,805,994 |
|
|
|
|
6,820,211 |
|
|
6,818,631 |
|
|
|
||||||||
|
Year Ended |
Nine months Ended |
|||||||||||||||||||
|
2020 |
2019 |
Increase/ |
2021 |
2020 |
Increase/ |
|||||||||||||||
|
Research and development costs |
|
|
|
|
|
|
|
|
||||||||||||
|
Drug manufacturing and testing |
$ |
170 |
$ |
623 |
$ |
(453 |
) |
$ |
47 |
$ |
105 |
$ |
(58 |
) |
||||||
|
Preclinical research |
|
281 |
|
106 |
|
175 |
|
|
123 |
|
202 |
|
(79 |
) |
||||||
|
Study IT-01 |
|
4,594 |
|
3,708 |
|
886 |
|
|
3,036 |
|
3,337 |
|
(301 |
) |
||||||
|
Study IT-02 |
|
— |
|
— |
|
— |
|
|
779 |
|
— |
|
779 |
|
||||||
|
Study IT-03 |
|
5 |
|
— |
|
5 |
|
|
434 |
|
— |
|
434 |
|
||||||
|
$ |
5,050 |
$ |
4,437 |
$ |
613 |
|
$ |
4,419 |
$ |
3,644 |
$ |
775 |
|
|||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
|
Research and development by |
|
|
|
|
|
|
|
|
||||||||||||
|
Salaries and payroll taxes |
$ |
1,101 |
$ |
782 |
$ |
319 |
|
$ |
1,132 |
$ |
745 |
$ |
387 |
|
||||||
|
Benefits |
|
212 |
|
149 |
|
63 |
|
|
183 |
|
156 |
|
27 |
|
||||||
|
Stock based compensation |
|
439 |
|
398 |
|
41 |
|
|
332 |
|
341 |
|
(9 |
) |
||||||
|
Clinical trial site costs |
|
1,119 |
|
1,481 |
|
(362 |
) |
|
1,180 |
|
799 |
|
381 |
|
||||||
|
Other costs from subcontractors and suppliers |
|
2,179 |
|
1,627 |
|
552 |
|
|
1,592 |
|
1,603 |
|
(11 |
) |
||||||
|
$ |
5,050 |
$ |
4,437 |
$ |
613 |
|
$ |
4,419 |
$ |
3,644 |
$ |
775 |
|
|||||||
45
|
Year Ended |
Nine months Ended |
|||||||||||||||||||
|
2020 |
2019 |
Increase/ |
2021 |
2020 |
Increase/ |
|||||||||||||||
|
General and administrative costs: |
|
|
|
|
|
|
|
|
||||||||||||
|
Salaries and payroll taxes |
$ |
234 |
$ |
206 |
$ |
28 |
|
$ |
190 |
$ |
183 |
$ |
7 |
|
||||||
|
Benefits |
|
18 |
|
16 |
|
2 |
|
|
14 |
|
13 |
|
1 |
|
||||||
|
Legal |
|
74 |
|
90 |
|
(16 |
) |
|
205 |
|
59 |
|
146 |
|
||||||
|
Patent and trademark |
|
167 |
|
110 |
|
57 |
|
|
62 |
|
130 |
|
(68 |
) |
||||||
|
Recruiting |
|
93 |
|
28 |
|
65 |
|
|
— |
|
79 |
|
(79 |
) |
||||||
|
Insurance |
|
59 |
|
51 |
|
8 |
|
|
55 |
|
44 |
|
11 |
|
||||||
|
Facilities and rent |
|
104 |
|
91 |
|
13 |
|
|
118 |
|
65 |
|
53 |
|
||||||
|
Investor relations |
|
133 |
|
211 |
|
(78 |
) |
|
167 |
|
112 |
|
55 |
|
||||||
|
Public relations |
|
64 |
|
167 |
|
(103 |
) |
|
6 |
|
61 |
|
(55 |
) |
||||||
|
Accounting services |
|
33 |
|
30 |
|
3 |
|
|
89 |
|
25 |
|
64 |
|
||||||
|
Other items less than 5% of total |
|
78 |
|
75 |
|
3 |
|
|
146 |
|
54 |
|
92 |
|
||||||
|
Stock-based compensation |
|
116 |
|
163 |
|
(47 |
) |
|
132 |
|
97 |
|
35 |
|
||||||
|
$ |
1,173 |
$ |
1,238 |
$ |
(65 |
) |
$ |
1,184 |
$ |
922 |
$ |
262 |
|
|||||||
Twelve Months Ended December 31, 2020 Compared to Twelve Months Ended December 31, 2019
Research and Development expenses increased primarily because we added research positions in June 2019, July 2019, and October 2020 and due to increased costs of our clinical trial as patient activity increased. 2019 includes the manufacture of a batch of INT230-6; costs related to the testing and shipment of this batch were incurred in 2020.
General and Administrative expense decreased. Legal expenses decreased since 2019 had higher costs related to the combination study agreements. Investor Relations decreased because we did not require an Investor Relations firm for most of the second half of 2020 since our next round of financing would not occur until the end of 2021. The increase in patent costs relates to the expansion into additional countries.
Nine Months Ended September 30, 2021 Compared to Nine Months Ended September 30 2020
Research and Development expenses increased due to increases in staffing and increases in patient enrolment in our clinical trial studies. In 2021, the Company planned and began treatment on study IT-02. In 2021, the Company also began the planning of study IT-03.
General and Administrative expenses include additional office space to accommodate our staffing increase. All other general and administrative costs were normal. In 2021, the increases are a result of increases in legal, accounting services, and other costs related to other professional services not directly related to the Initial Public Offering.
Liquidity and Capital Resources
Our financial statements have been prepared assuming we will continue as a going concern. The Company has incurred losses from operations and negative cash flows that raise substantial doubt about our ability to continue as a going concern.
Since our inception, we have not generated any revenue from product sales and have incurred significant operating losses. We expect to continue to incur significant expenses and operating losses for the foreseeable future as we advance the clinical development of our product candidates. We expect that our research and development and general and administrative costs will continue to increase significantly, including in connection with conducting clinical trials for our product candidates, developing our manufacturing capabilities and building and qualifying our manufacturing facility to support clinical trials and commercialization and providing general and administrative support for our operations, including the cost associated with operating as a public company. As a result, we will need additional capital to fund our operations, which we may obtain from additional equity or debt financings, collaborations, licensing arrangements or other sources.
46
We do not currently have any approved products and have never generated any revenue from product sales. We have financed our operations primarily through an initial investment from our founder and the issuance and sale of convertible debt notes, common stock and our convertible preferred stock to outside investors in private equity financings. From our inception through September 30, 2021, we raised an aggregate of approximately $32.1 million of gross proceeds from such transactions. As of September 30, 2021, our cash and cash equivalents were $7.4 million.
The following table summarizes the net cash provided by (used for) operating activities, investing activities and financing activities for the periods indicated (in thousands):
|
YEARS ENDED |
NINE MONTHS ENDED |
|||||||||||||||
|
2019 |
2020 |
2020 |
2021 |
|||||||||||||
|
Net cash (used in) operating activities |
$ |
(4,416 |
) |
$ |
(5,370 |
) |
$ |
(3,651 |
) |
$ |
(3,958 |
) |
||||
|
Net cash provided by (used in) investing activities |
|
1,697 |
|
|
4,564 |
|
|
(3,334 |
) |
|
— |
|
||||
|
Net cash provided by financing activities |
|
3,858 |
|
|
6,293 |
|
|
6,293 |
|
|
2,050 |
|
||||
|
Net increase (decrease) in cash and cash equivalents |
$ |
1,139 |
|
$ |
5,487 |
|
$ |
(692 |
) |
$ |
(1,908 |
) |
||||
Operating Activities
Net cash used in operating activities for the year ended December 31, 2019 was $4,416,345, primarily consisting of a net loss of $5,379,836 as we incurred expenses associated with our clinical programs. In addition, we had non-cash charges of $561,377 for stock-based compensation of options and warrants given to employees and consultants. Net cash used in operating activities was also impacted by the increase in un-invoiced patient care costs as enrollment expands.
Net cash used in operating activities for the year ended December 31, 2020 was $5,370,192, primarily consisting of a net loss of $6,030,480 as we incurred expenses associated with our clinical programs. In addition, we had non-cash charges of $555,488 for stock-based compensation of options and warrants granted to employees and consultants. Net cash used in operating activities was also impacted by the increase in accrued patient care costs as enrollment expands.
Net cash used in operating activities for the nine months ended September 30, 2020 was $3,650,999, primarily consisting of a net loss of $4,395,901 and the impact of non-cash charges for $512,470 of stock-based compensation and reduction in the right-of-use asset. Net cash used in operating activities was also impacted by an increase of liabilities, net of assets, primarily due to a $434,567 increase in accrued patient care.
Net cash used in operating activities for the nine months ended September 30, 2021 was $3,957,900, primarily consisting of a net loss of $5,494,482 as we incurred expenses associated with our clinical programs, and the impact of non-cash charges of $592,593 which were primarily for stock-based compensation. Net cash used in operating activities was also impacted by the increase in liabilities, net of assets, primarily due to an increase of $959,284 in accrued patient care.
Investing Activities
Net cash provided by investing activities for the year ended December 31, 2019 was $1,696,767, which was attributable to maturities of short term investments exceeding the purchase of short term investments.
Net cash provided by investing activities for the year ended December 31, 2020 was $4,564,813, which was attributable to maturities of short term investments exceeding the purchase of short term investments.
Net cash used in investing activities for the nine months ended September 30, 2020 was $3,333,938, which was attributable to the purchases of short term investments exceeding redemptions.
Financing Activities
Net cash provided by financing activities for the year ended December 31, 2019 was $3,858,660, consisting of the sale of Series C preferred stock.
Net cash provided by financing activities for the year ended December 31, 2020 was $6,292,633 consisting primarily from the sale of Series C preferred stock.
47
Net cash provided by financing activities for the nine months ended September 30, 2020 was $6,292,634, consisting primarily of the sale of Series C preferred stock.
Net cash provided by financing activities for the nine months ended September 30, 2021 was $2,050,000, consisting primarily of the issuance of a $2,000,000 convertible note.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules.
Seasonality
Our business experiences limited seasonality.
Contractual Obligations, Commitments and Contingencies
The following table provides our significant commitments and contractual obligations as of September 30, 2021:
|
Payments due by Period |
|||||||||||||||
|
($ in thousands) |
Total |
Less than |
1 – 3 Years |
4 – 5 Years |
More than 5 Years |
||||||||||
|
Office Lease |
$ |
384,621 |
$ |
190,217 |
$ |
194,404 |
$ |
— |
$ |
— |
|||||
JOBS Act Accounting Election
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Subject to certain conditions set forth in the JOBS Act, if, as an “emerging growth company”, we choose to rely on such exemptions we may not be required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404, (ii) provide all of the compensation disclosure that may be required of non-emerging growth public companies under the Dodd-Frank Wall Street Reform and Consumer Protection Act, (iii) comply with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis), and (iv) disclose certain executive compensation related items such as the correlation between executive compensation and performance and comparisons of the CEO’s compensation to median employee compensation. These exemptions will apply for a period of five years following the completion of our initial public offering or until we are no longer an “emerging growth company,” whichever is earlier.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or U.S. GAAP. The preparation of our financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our financial statements. These items are monitored and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base our estimates on historical experience, known trends and events, and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may materially differ from these estimates under different assumptions or conditions.
48
While our significant accounting policies are described in more detail in the notes to our financial statements included elsewhere in this prospectus, we believe that the following accounting policies are those most significant to the judgments and estimates used in the preparation of our consolidated financial statements.
Accrued Research and Development Expenses
Research and development costs are expensed as incurred. We record the estimated patient care costs as services are provided but not yet invoiced and include these costs in the accrued expenses in the balance sheet and within research other expense in the statement or operations.
Equity-Based Compensation
We recognize compensation costs related to stock option grants to employees and board members and warrant grants to nonemployees based on the estimated fair value of the awards on the date of grant. We estimate the grant date fair value and the resulting stock-based compensation expense using the Black-Scholes option-pricing model. The grant date fair value of the stock-based awards is recognized on a straight-line basis over the requisite service periods, which are generally the vesting period of the respective awards. Forfeitures are accounted for as they occur.
We historically have been a private company and lack company-specific historical and implied volatility information for our shares. Therefore, we estimate our expected share price volatility based on the historical volatility of publicly traded peer companies and expect to continue to do so until such time as we have adequate historical data regarding the volatility of our own traded share price.
Quantitative and Qualitative Disclosure about Market Risk
We are exposed to market risks in the ordinary course of our business. These risks primarily include interest rate sensitivities. Our interest-earning assets consist of cash, cash equivalents and short-term marketable securities such as US Treasury bills, which are denominated in U.S. dollars. We had cash, cash equivalents of $7,408,192, or 91% of our total assets, as of September 30, 2021. Interest, dividend, and investment income earned on these assets was $74,009 for the year ended December 31, 2020 and $2,261 for the nine months ended September 30, 2021. Our interest income is sensitive to changes in the general level of interest rates, primarily U.S. interest rates. Such interest-earning instruments carry a degree of interest rate risk; however, a change by 10% in interest rates would not have a material impact on our financial position or results of operations during the year ended December 31, 2020 and the nine months ended September 30, 2021.
We are not currently exposed to significant market risk related to changes in foreign currency exchange rates.
Inflation generally affects us by increasing our costs. We do not believe that inflation had a material effect on our business, financial condition or results of operations during the years ended December 31, 2019 and 2020 and the nine months ended September 30, 2020 and 2021.
Controls and Procedures
Historically, as a privately held company, we have maintained internal controls over financial reporting but we have a material weakness due to a lack of segregation of duties since we have a limited administrative staff. However, these internal controls have not been subject to the testing required under the standards of publicly traded companies by Section 404 of Sarbanes-Oxley. We are not currently required to comply with SEC rules that implement Sections 302 and 404 of the Sarbanes-Oxley Act, and are therefore not required to make a formal assessment of the effectiveness of our internal controls over financial reporting for that purpose. However, at such time as Section 302 of the Sarbanes-Oxley Act is applicable to us, we will be required to evaluate our internal controls over financial reporting.
49
Limitations on the Effectiveness of Controls
Our management, including the Chief Executive Officer and the Chief Financial Officer, recognizes that any set of controls and procedures, no matter how well-designed and operated, can provide only reasonable, not absolute, assurance of achieving the desired control objectives. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, with us have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of controls. For these reasons, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. This lack of segregation of duties from a limited number of administrative employees is a material weakness in internal controls. In August 2021, the Company engaged a Chief Financial Officer in order to add an additional layer of oversight on the financial reporting process and to reduce this material weakness.
50
Business Overview
Intensity Therapeutics, Inc. is a clinical stage biotechnology company passionately committed to applying scientific leadership in the field of localized cancer reduction leading to anti-cancer immune activation. Our new approach involves the direct injection into tumors of a unique product created from our DfuseRxSM discovery platform.
One challenge that we have identified with current intratumoral (IT) treatment approaches is that a tumor’s lipophilic, high fat and pressurized microenvironment does not effectively absorb water-based products. We believe that this drug delivery challenge limits the effectiveness of prior and current IT treatments that formulate their product candidates by injecting aqueous products (regardless of the mechanism or approach, i.e. the stimulation of an inflammatory response or efforts to attract immune cells into a hostile live tumor). Accordingly, there remains a continued unmet need for the development of direct IT therapies for solid tumors that provide high local killing efficacy coupled with nontoxic systemic anti-cancer effects. We believe we have created a product candidate with the necessary chemistry to overcome this local delivery challenge. Evidence shows the mechanism of tumor killing achieved by our drug candidate also leads to systemic immune activation in certain cancers.
Our platform creates patented anti-cancer product candidates comprising active anti-cancer agents and amphiphilic molecules. Amphiphilic molecules have two distinct structural components: one part of the molecule is soluble in water and the other is soluble in fat or oils. Certain amphiphilic compounds when mixed with medicinal agents allow those compounds to also become soluble in both fat and water.
Our lead product candidate, INT230-6, consists of two proven anti-cancer cytotoxic agents, cisplatin and vinblastine sulfate, mixed in water with the amphiphilic molecule 8-((2-hydroxybenzoyl)amino)octanoate (referred to as SHAO). The product is packaged in a single vial. The anti-cancer agents, cisplatin and vinblastine sulfate, used in our product candidate are both generic versions of the compounds. These agents are available to purchase in bulk supply commercially. The United States Food & Drug Administration (the “FDA”) previously approved both drugs as intravenous agents for several types of cancers. Cisplatin was first approved in 1978 for testicular cancer. Per the product labeling, cisplatin’s approved indications include treatment of testicular, ovarian and bladder cancer. The drug is also used widely in several other cancers including pancreatic and bile duct cancer. Vinblastine sulfate was approved in 1965. Per the product labeling, vinblastine sulfate’s approved indications include treatment of generalized Hodgkin’s disease, lymphocytic lymphoma, advanced carcinoma of the testis, and Kaposi’s sarcoma. The drug is also used in breast and lung cancer. In 2017, we initiated a Phase 1/2 dose escalation study with INT230-6 in the United States under an investigational new drug application (“IND”) authorized by the FDA and in Canada following receipt of a no objection letter from Health Canada. The study, IT-01, is exploring the safety and efficacy of INT230-6 in patients with refractory or metastatic cancers. We completed the Phase 1 dose escalation portion of this study. We are currently conducting the Phase 2 portion of the trial, which consists of several different expansion cohorts. One arm of the trial tests our drug candidate in a variety of cancers. Four of the cohorts combine our product candidate with Merck’s Keytruda® (pembrolizumab) and 3 arms combine our drug candidate with Bristol-Myers Squibb’s drug Yervoy® (ipilimumab). We are also evaluating INT230-6 in another Phase 2 study (the INVINCIBLE study) in Canada as a treatment for early stage breast cancer prior to surgery.
We believe that our drug candidate has achieved clinical proof-of-concept (POC). This means that there is sufficient availability of human clinical data confirming that the concept of a direct intratumoral injection in certain tumor types is feasible and that further investigation is reasonably likely to lead to drug approval and commercialization. We believe clinical POC has been achieved based on the broad range of data that we have generated from our preclinical experiments and clinical trials. Data shows that INT230-6 disperses widely throughout injected tumors, is absorbed well, penetrates and delivers the potent agents into tumor cells to kill them and activates a systemic immune response to fight the cancer. Our treatment approach utilizes intratumoral administration of INT230-6 to selectively induce tumor cell death and elicit an innate and adaptive anti-tumor immune response. Following injection of our product candidate, the tumors become highly necrotic, meaning that cancer cells die. After injection of INT230-6, tumors also become more amenable to immune cell infiltration. The tumor-killing process creates antigens, which are substances from the patient’s tumor that improve the recognition of the cancer by immune cells. While our product candidate is administered directly into the tumor, we have also observed in our preclinical studies and in our clinical trials that injections of INT230-6 can lead to a systemic immune response that attacks distal, uninjected tumors, a result known as an “abscopal” effect. Data generated in our trials show that our patented and patent pending drugs can extend life with less toxicity than is observed when these drugs are used by their normal route of administration, intravenously (IV).
Between the metastatic study IT-01 and the INVINCIBLE study we have treated over 115 patients as of September 30, 2021.
51
Our Pipeline
Our pipeline shown below is focused on realizing the full potential of INT230-6 in metastatic and local disease settings to help cancer patients with major unmet medical need. Together with our collaboration partners Merck and Bristol-Myers Squibb, we are exploring the use of our product candidate across multiple cancer types (including those types that do not normally respond to immunotherapy) and “hot” tumors (cancer types that are more likely to respond to immunotherapy). Within study IT-01 we are testing INT230-6 with Keytruda as treatment for pancreatic cancer, colon cancer, bile duct cancer and squamous cell cancer. Also, within IT-01 we are testing INT230-6 in combination with Yervoy for the treatment of sarcoma, breast cancer and liver cancer. In the Phase 2 randomized INVINCIBLE study, a collaboration with the Ottawa Hospital Research Institute, INT230-6 alone is also being used as treatment in early-stage breast cancer.
Based on survival data generated to date in sarcoma subjects, we have also designed a Phase 3 program to test our drug candidate in soft tissue sarcoma. Further, in 2018, we received Fast Track Designation by the FDA to use INT230-6 in metastatic triple negative breast cancer for patients whose cancer has progressed following one or two prior drug treatments. The FDA has reviewed our Phase 2/3 development plan for this indication. We plan to use the proceeds from this offering to initiate these Phase 3 programs.
We also continue to research and test new formulations for improved immune activating properties. Preclinical testing is in process, and we conducted a number of animal experiments and identified promising product candidates such as INT33X.

In addition, we plan to continue to research and test new product candidates with improved immune activating properties. Through research studies conducted in animals, we have identified a promising product candidate currently designated as INT33X. We believe that the INT33X product candidate development program will most likely lead to the creation of new patents and other intellectual property. As disclosed in the Use of Proceeds section of this prospectus, we anticipate spending a portion of the proceeds that we receive from this offering to conduct preclinical experiment work and fund the drafting of the new patents related to INT33X. As part of our development program for INT33X, we will first conclude our on-going research studies in mice, after which we will finalize the exact product candidate composition before proceeding with clinical development. We expect that we will be able to complete this work in 2022.
Our Strengths
• Experienced Oncology Pharmaceutical Development Management Team. Our CEO, Lewis H. Bender, has over 28 years of experience in the development of drugs using novel delivery technologies. While at Emisphere (a company purchased for $1.8 billion by Novo Nordisk in 2020), where he held many positions including CEO, Mr. Bender developed expertise in cGMP manufacturing, preclinical and clinical testing, biotech financing, quality assurance and drug development. He has taken products from research to Phase 3 testing and helped to create the commercial drug semaglutide branded as Rybelsus for type 2 diabetes that uses amphiphilic molecules to enhance diffusion of peptides across the gut. Mr. Bender also worked in manufacturing at Roche. Our Chief Medical Officer, Ian B. Walters. MD, has more than 20 years of clinical development experience and helped to test over 30 clinical compounds helped five products gain
52
FDA approval including the multi-billion dollar drugs Opdivo® and Yervoy® (trademarks of Bristol-Myers Squibb). He is a clinical and translational scientist with research experience at the Rockefeller University, Bristol-Myers Squibb and Millennium. Our Chief Financial Officer, James M. Ahlers, has over 25 years working building life science businesses. He has been CFO of Intarcia Pharmaceuticals and has raised capital through equity, debt and complex structured instruments, including IPOs. Our Sr. Vice President of clinical development, Syed Mahmood MD, has worked at large pharmaceutical (GSK, Novartis) and biotechnology (Progenics) companies. He helped launch multiple drugs including AZEDRA and PyL, and GSK’s/Novartis’s Tafinlar, Mekinist, Votrient, Luminespib and Buparlisib. Our Vice President of Project Management, Steve Innaimo, has over 20 years of experience from Bristol Myers Squibb, with expertise in all aspects of logistics and project management software. Our Vice President of Regulatory Affairs, Rebecca Drain also has over 20 years at Bristol Myers Squibb with more than 10 years of expertise in regulatory submissions. Our Principal Accounting Officer and Controller, John Wesolowski CPA has experience in public accounting at KPMG, with over 30 years of private and public controller experience including 18 years in the controller office at Yale University.
• Proprietary Drug Discovery platform, DfuseRxSM with Product Patent Protection in 37 Countries. Since our inception, we have conducted research using our discovery platform. Our technology platform allows us to identify novel product formulations and test the products’ activity in animal or test tube models of cancer. Using our platform technology, we have evaluated several formulations comprising various amphiphilic molecules that act as cancer cell penetration enhancers. We have tested formulations using our technology with many potent, anti-cancer drugs (with different mechanisms of action) in various combinations under several conditions to discover our lead product candidate. Our product candidates have a robust intellectual property position with 14 issued patents (3 issued in the US) and the ability to enforce our claims in 37 countries including the U.S. and all external major pharmaceutical markets. Five foreign patent applications are pending.
• Partnerships with World Leading Oncology Research Organizations and Major Pharmaceutical Companies
• The National Cancer Institute (NCI) Research Agreement. In May 2014, we were awarded a Collaboration Research and Development Agreement (CRADA) by the National Institute of Health’s National Cancer Institute. The CRADA was with the Vaccine Branch under the leadership of Dr. Jay Berzofsky. The research sought to understand the mechanism of action of INT230-6 and test the drug in several models in the NCI’s laboratories. The program resulted in a peer-reviewed publication titled Intratumorally delivered formulation, INT230-6, containing potent anti-cancer agents induces protective T-cell immunity and memory, which appeared in the journal OncoImmunology 2019 Vol 8 No 10; 15 and that was jointly authored by us and the NCI. The data for the paper was generated entirely by the NCI in their laboratories and reported the critical role of T-cells in promoting complete tumor regression using our drug candidate and that INT230-6 was synergistic with anti-PD-1 (programmed death receptor 1) and anti-Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) antibodies.
• Merck Partnership. In June 2019, we entered into an agreement with Merck to evaluate the combination of INT230-6 and KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 therapy, in patients with advanced solid malignancies, including pancreatic, bile duct, squamous cell and non-MSI high colon cancers. We dosed our first patient in this combination study in October 2019 and, through September 30, 2021, 21 patients in total have been dosed. After nearly two years of dosing a combination of Keytruda and INT230-6 we see comparable safety to INT230-6 monotherapy. In addition, there have been only a few low grade immune-related adverse events reported in the combination.
• Bristol Myers Squibb Partnership. In April 2020, we entered into a clinical trial collaboration agreement with Bristol Myers Squibb to evaluate the safety and efficacy of our product candidate INT230-6 with BMY’s CTLA-4 immune checkpoint inhibitor Yervoy® (ipilimumab). This combination is being evaluated in patients with breast cancer, liver cancer, and advanced sarcoma. Through September 30, 2021, we have dosed 14 patients in this combination arm. There have been low incidences of immune-related adverse events reported in the combination.
53
• Clinical Collaborations with World Leading Academic Hospitals in the US and Canada. To conduct our Phase 1/2 trial we have partnered with experienced clinicians at leading academic institutions in the US and Canada to test the safety and efficacy of our lead product candidate. The hospitals include Johns Hopkins Sydney Kimmel Cancer Center, Temple University’s Fox Chase Cancer Center, UMASS Memorial Hospital, The University of Southern California’s Norris & HOAG Hospitals, Columbia Presbyterian, the University of Toronto’s Princess Margaret Hospital and Houston Methodist.
• The INVINCIBLE study, with Canadian Centers of Cancer Research: the Ottawa Hospital Research Institute and the Ontario Institute of Cancer Research (OICR). In March 2021, we began this Phase 2 Randomized, Window of Opportunity trial evaluating clinical and biological effects of intratumoral INT230-6 against no treatment (the standard of care) in early stage breast cancer awaiting surgery. The primary endpoint is the proportion of patients who achieve a complete cell cycle arrest, defined as a reduction in the proportion of cells staining positive for Ki67, a widely used marker of cancer cell proliferation.
• Clinical Data Demonstrates the Anti-Cancer Activity in Humans in Multiple Cancers of Our Lead Product Candidate. INT230-6 has already generated solid evidence of activity as a single agent in clinical studies. Localized and abscopal effects have been observed in several patients. Tumor regressions with killing of the cancer cells is widely observed in injected lesions. Many patients who had exhausted all approved treatments for their type of cancer benefited from our product candidate. Our clinicians have reported tumor stabilization, tumor shrinkage, long periods without new tumors forming, a size reduction of un-injected tumors and a reduction in disease symptoms. These results are observed in combination with low toxicity over a period of several months and even well after our treatment has completed.
• Increased Survival Observed in Metastatic Disease. Preliminary data presented at ASCO 2021 indicates patients receiving INT230-6 appear to live longer compared to historical data for subjects in Phase 1 basket studies as reported in the literature.
• Acceptable Safety Profile of the New Drug/Treatment Approach to Date. As of September 30, 2021, there have been over 685 injections of INT230-6 into tumors, including 391 injections into visceral tumors deep in the body. Injection locations include the pancreas, liver, lung, and lymph nodes. No maximum tolerated dose has been reached. In our study IT-01 in metastatic patients most adverse events are minor grade 1 or 2; approximately 12% are grade 3 even when combined with other immunotherapies, with no grade 4 or 5 adverse events up to our recommended maximum doses. We believe the safety profile consists of mainly low grade related adverse events because the drug primarily stays in the tumor and the potent agents do not disperse throughout the body. Measurement of the amount of the drugs seen in the blood (pharmacokinetics or PK) indicates that more than 95% of the drug that is dosed remains in the tumor.
• Fast Track Designation from FDA for INT230-6 in Triple Negative Breast Cancer. On April 17, 2019, we announced that the FDA granted Fast Track Designation to our development program evaluating INT230-6 for the treatment of patients with relapsed or metastatic triple negative breast cancer (mTNBC) who have failed at least two prior lines of therapy. This important regulatory designation is based on the promising data observed to date from use of INT230-6 in our breast cancer research. This Fast-Track Designation (FTD) allows us to work more closely with the FDA to develop our new cancer treatment approach most effectively and efficiently for the indication reviewed by the FDA. The FTD may help us determine other potential indications or uses to pursue for INT230-6 in breast cancer.
• Phase 3 Program Sarcoma Study Designed and Discussed with FDA. Given the positive preliminary data on survival seen in our metastatic study in sarcoma patients, in August 2021 we requested a meeting with the FDA to discuss our Phase 3 program. The FDA granted us a meeting date and we met with FDA on October 14, 2021. We presented our existing sarcoma data and discussed our protocol synopsis. At the conclusion of that meeting, we believe there was alignment with FDA on the Phase 3 patient population, control groups and overall statistical design. We initiated drafting of the Phase 3 protocol based on that meeting. We have contacted contract research organizations to
54
help manage the trial. The Phase 3 trial can only begin after final review and approval of the protocol and other IND documents by FDA. We anticipate that the study could begin (assuming an acceptable FDA review and no disruptions) in the third quarter of 2022.
• A Results-Oriented Organization. We are committed to reporting data generated from our clinical trials and presenting such data at medical conferences. Data presentations made in Q4 2021 at important oncology conferences in 2021 included new results at the annual Society for Immunotherapy of Cancer (SITC) in 2021 (one presentation of INT230-6 alone or in combination with Keytruda and one presentation in combination with Yervoy in sarcoma), results in a highly refractory sarcoma patients at the Connective Tissue Oncology Society (CTOS) (selected for oral podium presentation) and results in metastatic breast cancer at the San Antonio Breast Cancer Symposium (SABCS). In the middle of 2022, we anticipate top line results from the INVINCIBLE study and new data from our clinical trial testing the combination of INT230-6 and Keytruda as well as the combination with Yervoy. We anticipate initiation of our Phase 3 programs as well as the selection of a next generation product candidate in 2022 (pending funding). Clinical manuscripts are in draft.
• A Company Focused on Reaching the Market with its Lead Product Candidate. We are focused. Our lead product candidate requires a significant amount of work to be completed including animal safety studies, toxicology studies, communication with regulatory authorities, manufacturing development, quality testing, assay development, manufacturing scale up, production, stability testing, and of course clinical trial testing from Phase 1 to 3. We are nimble and allocate resources with the objective of always generating the data necessary to advance our product candidate and technology platform forward under the best circumstances possible.
Our Strategy
We believe our approach may overcome some of the inherent problems of treating cancer with less toxicity. We intend to apply our deep understanding of novel drug delivery to create a range of new direct killing and immune-activating products candidates while focusing on our lead program. If successful, we hope to fundamentally change the way cancer is treated in multiple cancer types in both the metastatic and presurgical disease settings.
We seek to build a company that develops and commercializes a new medicine and treatment methodology. By applying a disciplined focus on product development, we seek to transform the lives of cancer patients and change the very essence of cancer treatment.
Our objective is for patients to overcome their cancer without harm, to live a long life with high quality and to eliminate the fear of disease recurrence. We maintain a culture of high integrity that embraces the patient and their caregivers. A simple strategy: taking care of the patient will benefit all stakeholders.
Key elements of our strategy include:
• Focus our resources to aggressively pursue the research and development of our novel medicine to transform patient lives.
• To always remember that taking care of and benefiting the patient is the most important element to being successful.
• Manage costs well by outsourcing research and development to qualified, academic, private or government laboratories or hospitals to leverage outside expertise while always maintaining our know-how, skill sets and intellectual property.
• Build an internal team of experienced industry veterans that can work independently and who know how to get the product development job done.
• Create a large body of rigorous data, publications, presentations, collaborations and training materials.
• Continuously find better methods to communicate to the medical community and patients of the power of our new approach.
• Continue our commitment to precision medicine and personalized care for each and every patient.
• Assure that our technology is fully understood, explored, and used as designed.
55
Market Opportunities for Our Product Candidates
The development of a tumor is a complex biological process involving uncontrolled cellular division and growth. Cancer arises from mutations in our own cells. When such cellular alterations happen the immune system often cannot distinguish between cancer and healthy cells. Cancer cells adapt to evade and thwart immune cells in several ways and can thus grow unchecked.
Cancer Statistics 2020, which is published in the American Cancer Society’s peer-reviewed journal, CA: A Cancer Journal for Clinicians, estimated that in 2020, over 606,000 Americans died from cancer. Cancer is the second most common cause of death in the U.S. after heart disease. According to the American Society of Clinical Oncology’s journal, the ASCO Post, the national cost of cancer care in the United States is expected to rise to $246 billion by 2030. As healthcare costs in general continue to escalate, expenses due to cancer are a major contributing factor.
Metastatic Disease
The overwhelming, unmet medical need is better treatment of solid tumors; 90% of cancer patient deaths are due to solid tumors. Unfortunately, even with the best new therapeutic agents, the long-term survival rates for inoperable or metastatic cancer are extremely low (often single digits) and toxicity (the collateral damage to the patient’s health) is debilitating.
Five-year Survival Percentage Rates for Metastatic, Late Stage Cancers
|
Cancer type |
5 Year Survival |
Cancer type |
5 Year Survival |
|||
|
Breast |
29 |
Ovarian |
30 |
|||
|
Colon/rectal |
15 |
Pancreas |
3 |
|||
|
Esophagus |
5 |
Prostate |
30 |
|||
|
Kidney |
14 |
Sarcoma |
16 |
|||
|
Larynx |
34 |
Testis |
95 |
|||
|
Liver |
3 |
Thyroid |
53 |
|||
|
Lung/Bronchus |
6 |
Urinary bladder |
6 |
|||
|
Melanoma (skin) |
30 |
Uterine cervix |
18 |
|||
|
Oral cavity |
40 |
Uterine corpus |
16 |
____________
* For cancers that have moved to distal sites
From data sources: Surveillance, Epidemiology, and End Results National Cancer Institute, SEER 5-Year Relative Survival Rates, 2011–2017
In late stage, metastatic disease, tumors often become resistant to all therapies, even after the agents have provided some efficacy benefit. The reality today for many cancer types is that if the disease is detected late, most treatments are highly toxic and few approaches provide patients with much hope of long term survival. Even with good outcomes, whether by surgical, chemical, radiative or ablation methods, the treatment is invasive, has severe side effects, damages the body and is mentally demanding on patients and their families.
Local Disease
Today, the annual number of interventional oncology procedures in the U.S. alone are estimated in the millions. For example, the majority of breast cancer tumors identified are local to the breast or are regional. As a result, there are 170,000 lumpectomies performed in the U.S. each year. Dr. Roshni Rao, Chief, Breast surgery program, at New York-Presbyterian/Columbia University Medical Center wrote in the Cancer Letter that “although lumpectomy is the best option for many breast cancer patients, with 170,000 procedures performed annually, it is not perfect. All too often, a post-operative pathology report shows that while the surgeon may have removed the entire tumor, a second surgical procedure is needed to clean up lingering cancer cells. Known as re-excision, it occurs in roughly 20% to 25% of cases, on average. It is critical for surgeons and their patients to have access to the latest innovations, once demonstrated effective by clinical research, be used wherever and whenever possible.” Our drug candidate’s potential to kill cancer quickly prior to surgery and engage an anti-cancer immune response may provide a higher percentage of patients a greater five year event-free survival for a number of tumor types.
56
Breast Cancer
About 1 in 8 U.S. women (about 13%) will develop invasive breast cancer over the course of her lifetime. An estimated 281,550 new cases of invasive breast cancer are expected to be diagnosed in women in the U.S. during 2021, along with 49,290 new cases of non-invasive (in situ) breast cancer. Breast cancer is the most commonly diagnosed cancer among American women. In 2021, it was estimated that about 30% of newly diagnosed cancers in women would be breast cancers. Breast cancer became the most common cancer globally as of 2021, accounting for 12% of all new annual cancer cases worldwide, according to the World Health Organization.
Approximately 11-17% of breast cancers test negative for estrogen receptors (ER), progesterone receptors (PR), and excess human epidermal growth factor receptor 2 (HER2) protein, qualifying them as triple negative. TNBC is considered to be more aggressive and have a poorer prognosis than other types of breast cancer, mainly because there are fewer available targeted medicines. Patients typically receive chemotherapy. According to a study published in the Journal of Clinical Oncology, patients who fail two lines of therapy for TNBC typically progress within nine weeks. Those who have failed three lines progress within four weeks.
Sarcoma
Soft tissue sarcoma is a broad term for cancers that start in soft tissues (muscle, tendons, fat, lymph and blood vessels, and nerves). These cancers can develop anywhere in the body but are found mostly in the arms, legs, chest, and abdomen. There are many types of soft tissue tumors, and not all of them are cancerous.
There are many types of sarcoma; however, the three most common are bone sarcoma (referred to as osteosarcoma), leiomyosarcoma and liposarcoma. Leiomyosarcoma is a type of sarcoma that grows in the smooth muscles. The smooth muscles are also in the hollow organs of the body, including the intestines, stomach, bladder, and blood vessels. In females, there is also smooth muscle in the uterus. When sarcoma is metastatic prognosis is poor; even with chemotherapy, half of people diagnosed with metastatic disease die within 15 months. Each year, 12,000 people in the U.S. are diagnosed with soft tissue sarcomas. About 3,000 patients have bone sarcomas.
Cancer Treatment
There is a high unmet medical need for improved cancer treatments. Currently, early detection coupled with surgery and systemic chemotherapy is the most effective treatment against most cancers. For metastatic disease, systemic chemotherapy represents the backbone of treatment for many cancers. However, chemotherapeutic resistance often results in therapeutic failure and eventually death. Not only is chemotherapy often ineffective for cancers that exhibit such resistance, this approach is also highly toxic for many patients (Cancer Cell Int. 2015; 15:71). Almost all of the current anti-cancer drug therapies load drug throughout the entire body including classic chemotherapy before surgery (neoadjuvant), after surgery (adjuvant), targeted therapy, antibodies or antibody drug conjugates, liposomal or nanoparticle delivered drugs. Many cancer cells in tumors are located away from blood vessels (referred to have hypoxic regions) and systemic administration is ineffective at delivering the needed amounts of the medicine to all parts of the tumor. Thus, a significant limitation of the current anti-cancer treatments is proper drug delivery. Another challenge for systemic approaches is poor absorption or cellular mechanisms in the cancer cell to remove the drugs.
Immunotherapy
There has been much media and news reporting about the promise of immunotherapy in treating cancer. These novel product candidates are designed mobilize an immune system to attack the cancer. The field of cancer immunotherapy has become the primary focus of treatment for many tumor types. There is significant interest from pharmaceutical companies, physicians and patients in advancing new, immune-based treatment concepts. For the first time, some patients having a variety of formerly fatal cancers are experiencing long term survival benefit. Immunotherapy has shown promise against the most mutated cancers such as melanoma, renal cell carcinoma, squamous cell carcinomas and a subset of lung cancers. Often these new immune stimulating drugs work in patients having high levels of specific markers, such as the percentage of a protein on the surface of the cancer cell known as PD-L1 or the number of genetic mutations that may have caused the cancer referred to as a tumor’s mutational burden.
Unfortunately, immunotherapy has not worked well for the majority of solid tumor types, including the deadliest cancers such as sarcoma, pancreatic cancer, of the majority of colon cancers, triple negative breast cancer and brain cancer. Sometimes when using immunotherapies, the immune system has trouble distinguishing cancer from normal
57
tissue and attacks healthy cells. Thus, the immune therapies induce side effects. To enable more patients to benefit from immunotherapy, new technologies that are able to improve recognition of the cancer by the immune system, or disrupt the tumor’s ability to evade immune cells, are critical and strongly needed.
Challenges Facing Current Treatments
We believe that an effective cancer treatment must overcome three major problems.
1. The diverse nature of the disease: In most patients, there are two populations of the cancer with different physical properties. The local component is comprised of the well-defined, visual large tumors, seen in x-ray or imaging scans, that invade organs and tissue. The systemic aspect is comprised of cells circulating or implanted throughout the body. Essentially cancer is often simultaneously both microscopic (unseen) and macroscopic (seen).
2. Unreachable parts of tumors: Current methods of delivering cancer drugs either orally or intravenously (IV) do not reach many portions of tumors due to a lack of blood supply. These areas are referred to as hypoxic (low oxygen) regions. These areas of the tumor can also impede the influx of immune cells. IV or system dosing of cytotoxic agents suppresses the systemic immune system (Mathios et al, STM 2016) and reduces the potential of immunotherapies.
3. Lack of immune cell recognition and activation by tumor processes to evade: Immune cells have difficulty recognizing/distinguishing cancer cells from normal cells. Cancer also can cloak itself from the immune cells and create barriers to reduce their influx into the tumor.
Our Treatment Approach
Our treatment concept pioneers a new approach to treating cancer — kill tumors in the body (in situ) to create from the patient’s own cancer a recognizable, high-quality material (referred to as antigen) for better immune cell engagement against the cancer.
Our new treatment concept is to diffuse potent drugs throughout the tumor to saturate the tumor with strong killing agents. The active agents themselves used in our product candidate also have properties that improve immune recognition of the cancer. At the right dose our product candidates can completely saturate an injected tumor delivering high concentrations of drug into the cancer cells and killing the tumor. This process removes the cancer’s cloaking system, decreases the barriers to immune influx and activates a body-wide anti-cancer immune response to attack the uninjected tumors and unseen metastases. Our clinical data suggests that not all tumors need be injected for long term disease control.
Through our novel, drug treatment technology, we hope to transform the lives of patients with cancer. Our objectives are to increase patient longevity, reduce side effects, remove the fear of treatment, empower the patient, and minimize the risk of disease recurrence.
INT230-6, Our Lead Product Candidate
Our product candidate, INT230-6, consists of two proven anti-cancer cytotoxic agents mixed with a penetration enhancing amphiphilic molecule (SHAO), the chemical structure of which is shown in figure 1 below. When injected into tumors, INT230-6 can kill the tumors. Our safety studies show that if the drug is (accidently) injected into healthy tissue there is no observation of damage. The drug agents enter the blood stream at low doses. The unique amphiphilic SHAO compound formulated product candidate increases the dispersion of the drug throughout the tumor following intratumoral injection. Our technology is novel and unique. For those familiar with drug delivery technologies in cancer, it is important to note that our product candidate is not a liposome, not a nanoparticle nor an emulsion. INT230-6 is a 100% water-based formulation with tissue dispersion properties that do not destroy cancer cell membranes. We are not aware of any previous anti-cancer drug or prior intratumoral preparation with similar characteristics.
58
Figure 1 chemical structure of SHAO
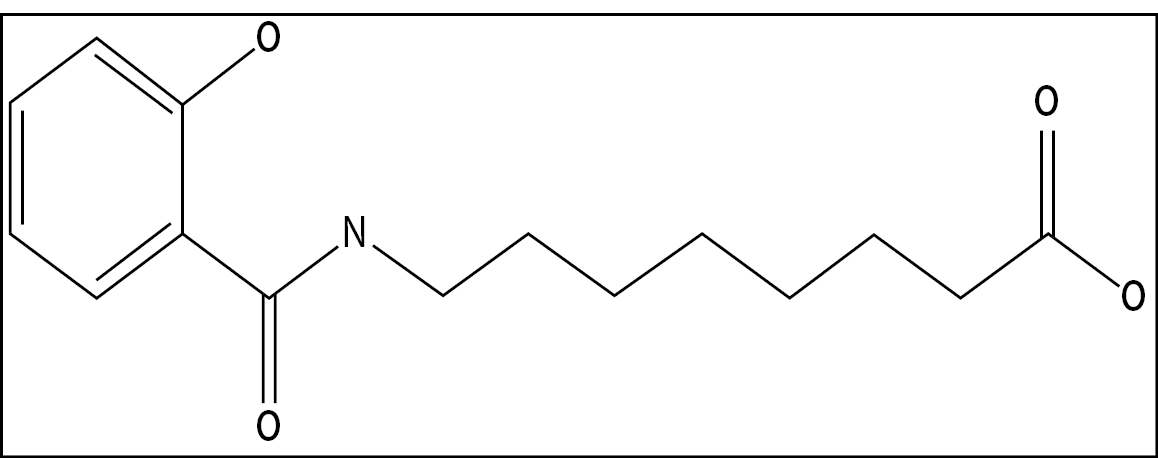
The SHAO molecule facilitates drug dispersion throughout the tumor. The molecule allows the tumor to absorb the killing agents and facilitates their diffusion into the cancer cells. Once in the cancer cell one drug cisplatin binds the DNA and causes the cell apoptosis (death) whereas the other agent, vinblastine sulfate, destroys the cell’s tubulin to shut down replication. Data in humans suggests that when administered at the proper drug dose to tumor volume ratio, a significant portion of the injected tumor can be killed on a single dose. In addition, there is evidence (in both animals and humans) that for certain cancers there is an activation of the immune system.
Our intratumoral (IT) technology is different than other IT approaches in four important ways:
1) We recognized that the composition of a tumor is highly unfavorable to direct injection of water-based products because the tumor has a high fat content and is under surrounding pressure. To be effective, an IT drug must disperse, be absorbed by the tumor and enter the cancer cell. Without our unique formulation chemistry water soluble drugs are not readily dispersed or absorbed by a tumor.
2) Our delivery technology is based on a proven science that uses amphiphilic molecules to transport drugs through tissue. The active drug agents in our lead product candidate (cisplatin and vinblastine sulfate) are established, commercial, potent killing agents with immune stimulating properties that as of now are only sold as IV products. Both cisplatin and vinblastine sulfate have dual direct killing and immune activating mechanisms of action. Cisplatin binds to DNA to cause apoptotic cell death and also attracts and binds T-cells via TL9 receptors. Vinblastine sulfate destroys tubulin to stop replication and also induces dendritic cell maturation.
3) Unlike other IT products, our product candidates have multiple opportunities well beyond skin tumors, such as melanoma. Our lead product candidate, INT230-6, has shown the ability to kill tumors deep in the body such as in the liver, lung, and peritoneum. The product candidate has also demonstrated ability to kill tumors from several cancer types with abscopal effects and increased overall survival.
4) Our product candidate has potential to kill tumors quickly and could be used before surgery immediately after diagnosis or for treatment of cancers where there are no therapeutic agents or suitable local treatments available.
INT230-6 in Animals
Our first research studies in mice were conducted with organizations that provide services under contract, referred to as contract research organizations (CROs). Our Company collaborated with the Vaccine Branch of the National Cancer Institute (NCI) in Bethesda, MD. The research with the NCI was established after the National Institutes of Health awarded our Company a cooperative research and development agreement (CRADA). The program was quite successful and culminated with the publication of a paper in July 2019 that we jointly authored with the NCI. In that publication we reported that INT230-6 treatment resulted in regression from baseline in 100% of the tumors and complete response in up to 90%. Experiments showed a critical role of T-cells in promoting complete tumor regression. Mice with complete response were protected from subcutaneous and intravenous re-challenge of cancer cells. Thus, immunological T-cell memory was induced by INT230-6.
As part of our own research, we formulated cisplatin in water without the SHAO and added a noncolloidal dye. When injected into a human pancreatic tumor grown in a mouse model, we observed that the water formulation of the drug without the SHAO was not absorbed in the tumor. The liquid mostly leaked from the tumor. However, the formulation that incorporated SHAO was readily and rapidly absorbed by the tumor in a dose dependent manner as shown in figure 2 below.
59
Figure 2 Comparison of drug dispersion/absorption in tumors with and without our DfuseRx technology.
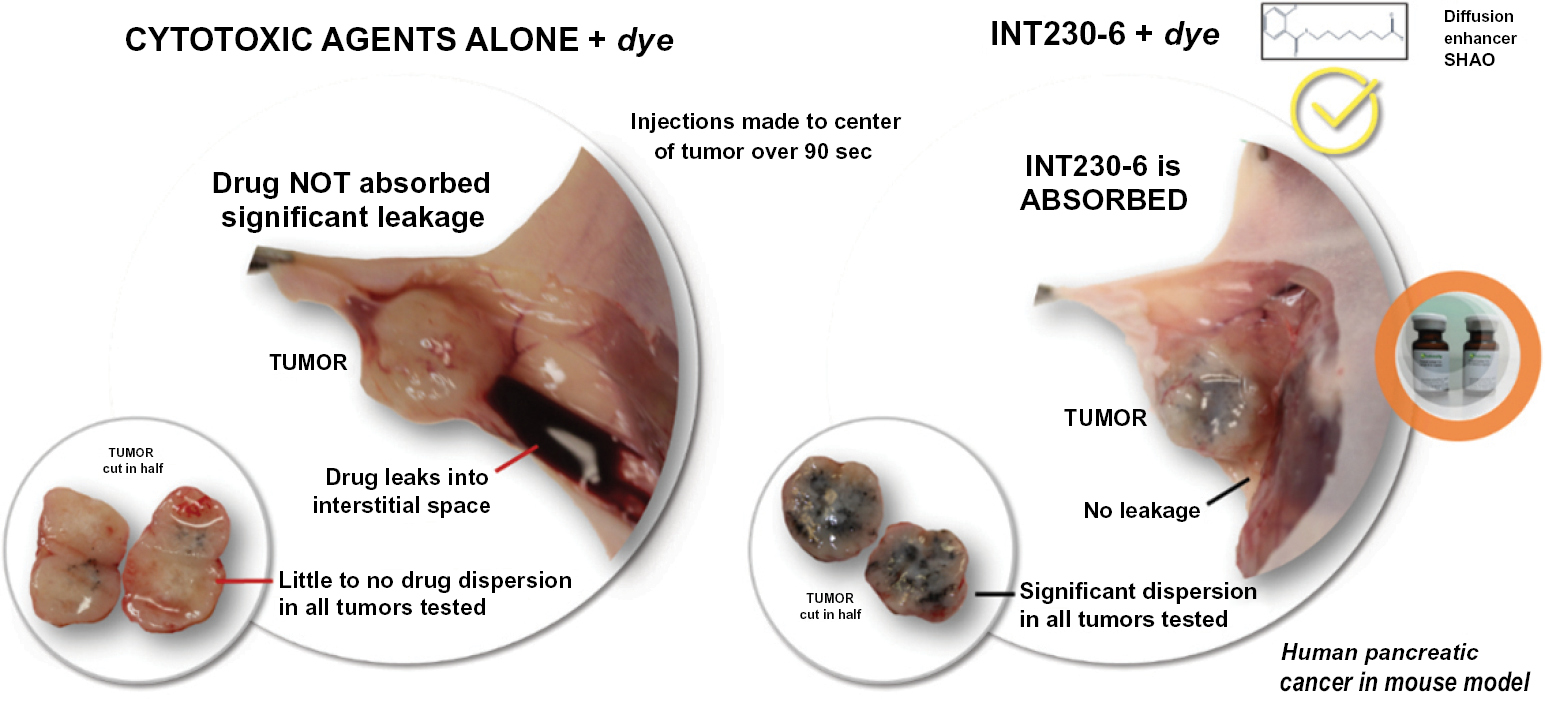
Dense human pancreatic cancer BXPC-3 tumors were grown in severe combined immunodeficiency mice. Injections using a metered pump of the cisplatin with dye in water were compared to INT230-6 with dye. Fourteen mice were treated. INT230-6 is well absorbed and distributed throughout tumors (right side images) compared to the drug alone in water which leaks out (left side images). Data published in the International Journal of Molecular Sciences June 2020 doi.org/10.3390/ijms21124493.
In addition to formulation experiments we conducted growth inhibition experiments using large tumors (>300 mm3) and treated with low drug doses. Typically, research conducted by other companies developing cancer products use small tumors (25 to 100 mm3). Such companies also often use large drug doses in their studies with drug amounts that are five to fifty-fold above our dose amounts. Our product candidate can completely eradicate murine tumors, an effect that is termed a complete response (CR). Most competitors show only a slowing down of the tumor’s growth rate over time.
INT230-6 regresses tumors over time as shown in figure 3 panel A and extends animal life compared to the drugs given alone intratumorally at the same dose without our technology. In addition, our drug candidate shows superior efficacy given intratumorally compared to dosing the drugs intravenously. Often animals with a CR are permanently protected against the cancer. This means upon re-inoculation with the same cancer new tumors do not grow. The protective effect happens whether the cancer cells are reinoculated under the skin or administered intravenously indicating a broad systemic immune protection.
Through our research collaboration with the NCI, we generated data regarding the mechanism of action for our lead product candidate. INT230-6 shows direct tumor killing and immune cell activation. The direct tumor cell death is caused by action of the two potent agents (cisplatin and vinblastine sulfate). Data generated to date indicates infiltration of dendritic cells into the tumor which can present antigen to activate CD8 and CD4 immune T-cells against the cancer. Survival and tumor eradication are mostly driven by CD8+ T-cells. Thus, our product candidate generates high quality, vaccine-like antigen from the attenuated tumors to promote the immune activation. The Company also published data showing increases in dendritic cells, macrophage, T-cells and Natural Killer (NK) cells 10 days after intratumoral treatment in mouse colon tumors. Selective immune depletion of CD4 and CD8 abrogates the therapeutic effect. Figure 3 panel B that shows the influx of various immune cell into the tumor microenvironment.
The scope of the NCI studies was to assess growth inhibition, survival and immune activation. Naïve mice were SC challenged with 1 × 106 C26 cells into the right flank. Vehicle or INT230-6 (0.5 mg/ml cisplatin, 0.1 mg/ml vinblastine sulfate, 10 mg/ml IT-006 cell penetration enhancer) were intratumorally (IT) administered into 300 mm3 (approx. 8.5 mm in diameter, 100 μl/400 mm3 C26 tumor) SC tumors (n = 10/group) for 5 sequential days (day 0 to 4) and tumor growth was monitored. The fraction 5/10 indicates the number of complete responders. The log rank test indicates a significant difference between the groups (p<0.0001).
60
Figure 3 Mouse data showing tumor reduction and immune activation
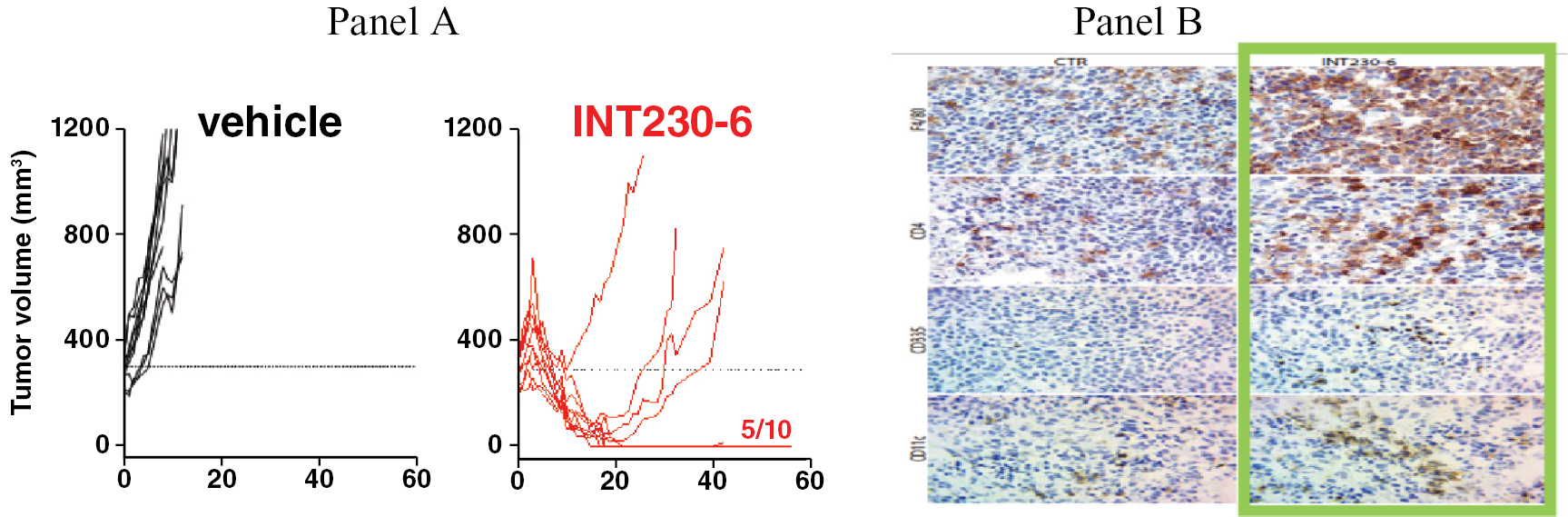
In Panel A on the left, 100% of animals receiving INT230-6 treatment for 5 days have a slight increase followed by a decrease from baseline, with 50% of animals having a complete response compared to no treatment controls with no decrease or complete responders (data generated by the NCI see OncoImmunology 2019 Vol 8 No 10; 15). Panel B cell staining shows an increase in the immune infiltrates. Data from Int. J. Mol. Sci. 2020, 21, 4493.
INT230-6 is Synergistic with Checkpoint Blockade
Nature has created checkpoints on the immune system to regulate the activity of the immune cells. These pathways are crucial for self-tolerance to prevent the immune system from attacking healthy cells indiscriminately. Large pharmaceutical companies such as Merck, Roche, AstraZeneca, Pfizer and Bristol Myers Squibb (BMS) have developed new types of anti-cancer anti-body drugs with the ability to modify and block the checkpoints on the immune system.
Our results show strong benefit in regressing tumors with the combination of INT230-6 and checkpoint inhibitors which leads to improve survival. The data showed the combination of our product candidate with either anti-PD-1 or CTLA-4 antibodies in a dual tumor (metastatic) cancer mouse resulted in additive benefit. The data was generated by our partners at the National Cancer Institute and under our CRADA and published (OncoImmunology 2019 Vol 8 No 10; 15).
Preclinical Good Laboratory Practice (GLP) Safety of INT230-6
During a pre-meeting in 2014 with FDA we reached agreement on an accelerated safety and manufacturing program. We successfully completed the needed tasks to begin clinical testing that included conducting pharmacology studies (showing activity of the drug), toxicology studies in two animal species, analytical methods development, manufacturing scale up, and regulatory submissions. All these steps were completed by 2015. The data showed that the use of SHAO did not change or increase the toxicity of cisplatin or vinblastine sulfate. Analytical results showed the two drugs remain unchanged chemically when INT230-6 is stored properly, which is in a standard freezer at -20°C.
Pre-Clinical Regulatory Interactions
In the United States, the U.S. Food and Drug Administration (FDA) regulates drug and device products under the Federal Food, Drug, and Cosmetic Act and its implementing regulations. The primary mode of action for our product candidate is expected to be attributable to the two drug components. Since our product candidate consists of small molecules, the FDA’s Center for Drug Evaluation and Research has primary jurisdiction over our product candidate’s pre-market development and review. Please see the section entitled “Risk Factors” for a description of some of the uncertainties regarding the timing or outcome of the regulatory approval process relating to our technology. We have been working closely with the Division of Oncology 1 (DO1), which is responsible for breast, gynecologic, and genitourinary cancers. We are working with the Division of Oncology 3 (DO3) to conduct a Phase 3 clinical trial of INT230-6 in patients with sarcomas.
As noted above, we met formally with the FDA (DO1) in a pre-investigational new drug application (pre-IND (as defined below)) Type B meeting in August 2014 and then completed the agreed upon preclinical safety program on INT230-6. We filed our investigational new drug application (“IND”) application and held a meeting with senior FDA officials in November 2016. In December 2016 FDA provided us a “Study May Proceed” letter.
61
We also met formally with the Canadian regulatory agency Health Canada (HC) in a preclinical trial application (CTA) meeting in 2016. We filed the CTA and held meetings with senior HC officials. Health Canada provided us a “No Objection” letter in early 2017. As we have progressed our study, we filed several amendments since 2017 and have received “No Objection Letters” each time from Health Canada. We have been treating patients continuously under both our IND and CTA since May 2017.
The regulatory agencies agreed to permit setting the drug dose based on tumor volumes rather than using alternatives such as dose based on a patient’s height and weight. Our belief is that using the patients’ total tumor burden instead of body size is a more personalized and precise approach to ensure that patients receive an appropriate dose for their unique cancer burden. Better dosing could lead to maximized efficacy with minimized side effects. In our clinical trial, tumor volume is calculated from radiographic imaging on target tumors at baseline. Dose for a given tumor is set based on its size.
IT-01 Phase 1/2 Clinical Trials
Our first study (IT-01), which began in 2017, is entitled “A Phase 1/2 Safety Study of Intratumorally Administered INT230-6 in Adult Subjects with Advanced Refractory Cancers”. The study design permits our product candidate to be tested in several different cancer patient populations with dosing into both superficial e.g. squamous cell, thyroid, breast, head and neck, lymphoma, melanoma and deep body tumors such as those found in pancreatic, liver, colon, bile duct, sarcoma, and chordoma cancers. The clinical trial seeks to determine the safety and potential efficacy of dosing INT230-6 directly into several types of cancers. We have tested our product candidate in 19 different tumor types. The Phase 1 portion of the study completed in July 2020.
Study, IT-01, is listed on clinicaltrials.gov; NCT03058289. The hospitals currently enrolling patients in the United States are: the Sydney Kimmel Cancer Center at Johns Hopkins, The Fox Chase Cancer Center at Temple University, the Norris Cancer Center, LA County and HOAG Presbyterian Hospitals of the University of Southern California medical system, the UMASS Memorial in Worchester Massachusetts, Columbia Presbyterian in New York, the Princess Margaret Hospital which is part of the University Health Network in Toronto, and the Houston Methodist.
Since the initial regulatory submissions, we have made six amendments to the IT-01 protocol. The most recent amendment simplified setting dose and eliminated blood collection for pharmacokinetic evaluation as dose escalation was completed and focus is now on the Phase 2 expansion cohorts including 4 cohorts with Merck’s Keytruda® (pembrolizumab) and 3 cohorts with Bristol Myers Squibb’s Yervoy® (ipilimumab). A schematic of the Phase 2 study’s three on-going cohorts is shown in figure 4 below.
Figure 4 current schema of the 3 on-going cohorts in the metastatic study IT-01.
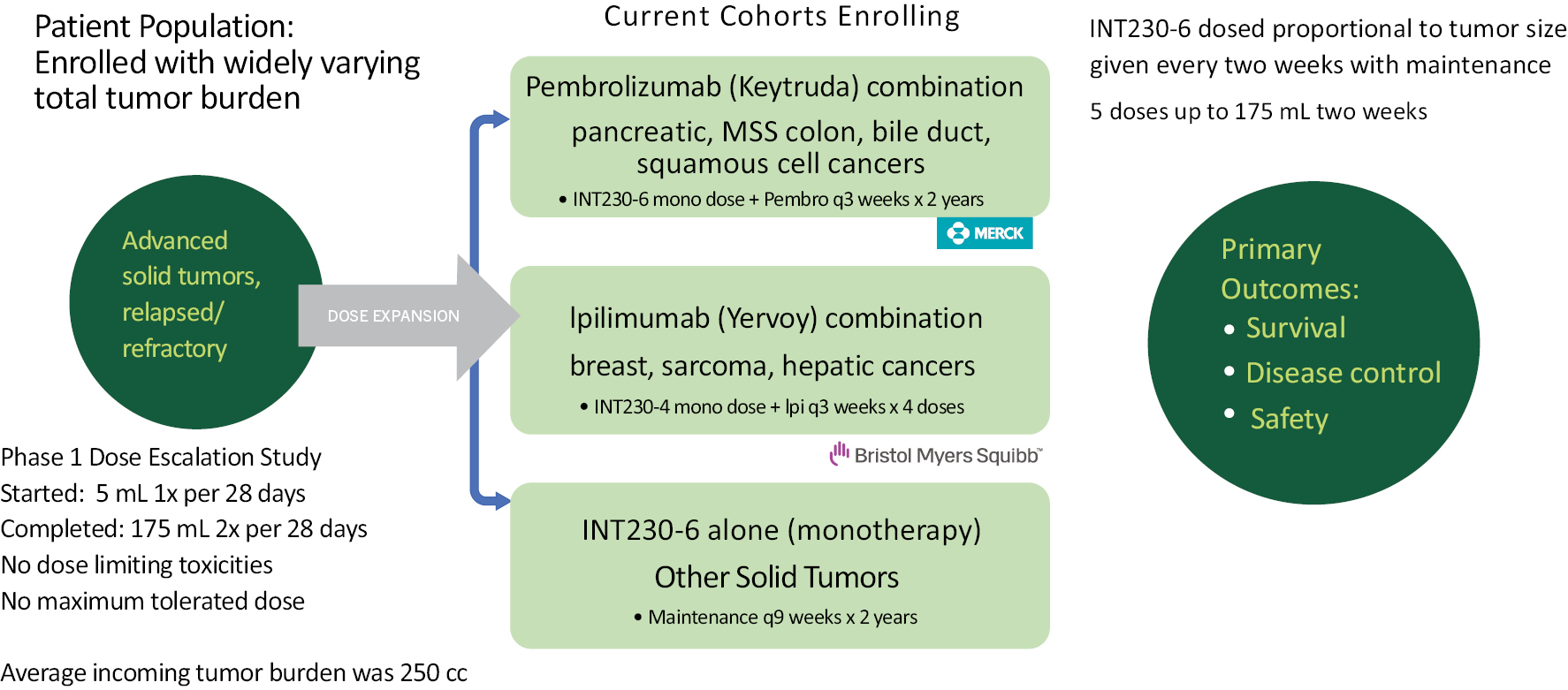
62
Clinical Collaborations
Merck Partnership
On June 25, 2019, we announced entering into an agreement with Merck Sharpe and Dohme to evaluate the combination of our lead product candidate INT230-6 and KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 (programmed death receptor-1) therapy, in patients with advanced solid malignancies including pancreatic, bile duct, squamous cell and non-MSI high colon cancers. Keytruda annual sales in 2020 are tracking to exceed over $12 billion. INT230-6 is highly synergistic with anti-PD-1 antibodies in mice. Survival and tumor response are significantly increased using the combination. One squamous cell carcinoma patient in our monotherapy study, who was recommend for arm and shoulder amputation prior to beginning INT230-6, received 4 doses of INT230-6 from March to July of 2018. That patient had a following treatment with pembrolizumab, (outside of our study) and subsequently had a complete response.
On October 31, 2019, we announced that the first patient has been dosed with a combination of INT230-6, and Keytruda. The combination is being studied in a series of cohorts within our ongoing Phase 1/2 international clinical study (NCT03058289). On March 30, 2020, we announced successful completion of the safety lead in portion of the IT-01 KEYNOTE A10 study arm (NCT03058289) that is testing the combination for safety. The cohort treated seven patients with different types of advanced cancers that were amenable to superficial injections including triple negative breast cancer (n=3) Merkel cell carcinoma, chordoma, desmoid tumor, and soft tissue sarcoma. Patients’ tumors were treated every two weeks for 5 doses with INT230-6 in combination with 200 mg of Keytruda every three weeks. All seven patients completed the 28-day dose limiting toxicity (DLT) evaluation period with no DLT’s or drug-related serious adverse events. The safety profile appears to be similar to INT230-6 monotherapy. Following completion of the dosing of INT230-6, patients continued Keytruda monotherapy for up to 2 years. Scans were collected regularly on patients to evaluate the efficacy of the combination. In the study cohort dosing INT230-6 with Keytruda as of October 20, 2021, there was a total of two treatment related adverse immune events reported. Both low grade related adverse events were in a different patient and attributed to Keytruda. There was one episode of a gamma glutamyltransferase increase and one hypothyroidism event. Both patients recovered.
The study steering committee, which is comprised of the principal investigators, reviewed the safety data and approved dosing into deep tumors and authorized initiation of the Phase 2 studies. The KEYNOTE A10 Phase 2 studies are enrolling patients with pancreatic cancer, microsatellite stable colorectal cancer and cholangiocarcinoma. These cancers are typically immunologically cold and historically non-responsive to immunotherapies. We also plan to test the combination in squamous cell carcinoma patients who have already failed a PD1/PDL1 agent.
Pursuant to our agreement with Merck, we are the sponsor of the clinical trial and are responsible for the costs of conducting it, and Merck will supply KEYTRUDA® for use in the clinical trial at no charge to us. The agreement does not provide for any milestone payments, royalties or other compensation to be paid to either party. The agreement provides for joint ownership of any inventions, clinical data and results solely generated in the combination portion of the clinical trial that relate to the combined use of the two drugs. Merck will solely own any inventions generated in the clinical trial that relate solely to KEYTRUDA® and all data resulting from testing performed by third parties engaged by and on behalf of Merck for some samples collected during the clinical trial. We will solely own any inventions generated in the clinical trial that relate solely to INT230-6, clinical data resulting from the use of INT230-6 as a monotherapy, and from all data resulting from testing performed by or on behalf of us on samples collected during the clinical trial. The term of the agreement will continue until delivery of the final report for the clinical trial, provided that either party may terminate the agreement due to the other party’s uncured material breach, a violation of anti-corruption obligations, patient safety concerns, regulatory action that prevents supply of such party’s compound, or such party’s termination of its compound’s development or withdrawal of its compound’s regulatory approval. Merck may also terminate the agreement if we fail to make any changes to the clinical trial protocol regarding the use of KEYTRUDA® that are requested by Merck in good faith to address any concern raised by Merck that KEYTRUDA® is being used in the clinical trial in an unsafe manner.
Bristol Myers Squibb (BMS) Partnership
On April 14, 2020, we announced that we had entered into a clinical trial collaboration agreement with Bristol Myers Squibb Company. The program evaluates the safety and efficacy of our lead product INT230-6, in combination with Bristol Myers Squibb’s Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) immune checkpoint inhibitor Yervoy® (ipilimumab). The combination is being evaluated in patients with breast cancer, liver cancer and advanced sarcoma in Phase 2 cohorts within IT-01, our ongoing Phase 1/2 clinical trial. We sponsor and conduct the clinical trial and Bristol Myers Squibb supplies Yervoy for use in the study. Yervoy is an immunotherapy
63
approved in melanoma and in combination with nivolumab in other indications as well. The most common severe immune-mediated adverse reactions from Yervoy (ipilimumab) are enterocolitis, hepatitis, dermatitis (including toxic epidermal necrolysis), neuropathy, and endocrinopathy. The majority of these immune-mediated reactions initially manifested during treatment; however, a minority occurred weeks to months after discontinuation of Yervoy.
Yervoy sold nearly $510 million in the second quarter of 2021 and primarily treats melanoma or kidney cancer. In 2018, Dr. James Allison won the Nobel Prize for recognizing that a protein on immune cells named CTLA-4 stopped immune cells from fighting cancer. Dr. Allison helped develop Yervoy to block CTLA-4 and unleash immune cells against cancer. Yervoy is quite potent, but has a relatively high percentage of severe side effects. We believe that improved recognition of cancer by the immune system, which our product candidate enables, may reduce the toxicities of the immune regulating agents and boost efficacy.
In the Yervoy combination arm of our trial, INT230-6 injections are given every 2 weeks for 5 sessions at fixed maximal dose into superficial or deep tumors, unlimited number of tumors to be treated per session with retreatment once every 9 weeks for two years. Yervoy (ipilimumab) is dosed concurrently starting at Day 1 every 3 weeks for four treatments for selected cancer types breast, liver and sarcoma. The types of cancers being evaluated in the combination arm of our study with Yervoy are sarcoma, liver and breast cancers. We are comparing our drug candidate alone to the combination in each of the cancer types. As of September 30, 2021, there have been 5 Yervoy related immune adverse events in the combination in a total of 4 patients. The events were colitis (1), hepatitis (1), and 3 episodes of rash maculo-papular in 3 different patients.
Pursuant to our agreement with BMS, we will sponsor, conduct and fund the Phase 1/2 trial, and BMS is obligated to supply Yervoy to us for no cost. The agreement does not provide for any milestone payments, royalties or other compensation to be paid to either party. The agreement provides for joint ownership of any inventions, clinical data and results solely generated in the combination portion of the clinical trial that relate to the combined use of the two drugs. BMS will solely own any inventions generated in the clinical trial that relate solely to Yervoy and all data resulting from testing performed by third parties engaged by and on behalf of BMS for some samples collected during the clinical trial. We will solely own any inventions generated in the clinical trial that relate solely to INT230-6, clinical data resulting from the use of INT230-6 as a monotherapy, and from all data resulting from testing performed by or on behalf of us on samples collected during the clinical trial. After the completion of the Phase 1/2 trial, we are obligated to provide BMS with a final report of the data resulting from the trial. Our agreement with BMS will terminate upon the completion of the Phase 1/2 trial, the delivery of a final report containing the data resulting from the combination cohort of Yervoy and INT230-6. Either party may terminate the agreement upon a material breach by the other party that remains uncured following written notice of such breach or upon certain bankruptcy events. In addition, either party may terminate the agreement immediately upon written notice if such party reasonably deems it necessary in order to protect the safety, health or welfare of subjects enrolled in the Phase 1/2 trial, or if a clinical issue arises with respect to INT230-6 or Yervoy that adversely impacts the ability to conduct the Phase 1/2 trial.
IT-02 (The INVINCIBLE Study)
In March 2021, we began this Phase II Randomized, Window of Opportunity trial evaluating clinical and biological effects of intratumoral INT230-6 against no treatment (the standard of care) in early stage breast cancer patients awaiting surgery. The primary endpoint is the proportion of patients who achieve a complete cell cycle arrest, defined as a reduction in the proportion of cells staining positive for Ki67, a widely used marker of cancer cell proliferation. The study design is shown in the figure 5 below. According to our estimates using the National Cancer Database (NCBD), approximately 39% of diagnosed with breast cancer nearly 100,000 patients have no therapeutic treatment following diagnosis. Women undergoing surgery typically wait 3 to 5 weeks to have the procedure.
The trial is a Phase II, randomized, open label, multi-center study to enroll up to 90 patients with early-stage breast cancer. Patients, randomized 2:1 to treatment, will receive either three doses of INT230-6 on days 1, 8 and 15 post diagnosis or no treatment, the current standard of care (SOC) prior to resection. The Study shall be conducted under the direction and supervision of Principal Investigator, Dr. Angel Arnout. Dr. Arnout will perform all those responsibilities assigned to principal investigators for personal performance by applicable Health Canada (HC) regulations. The study shall evaluate the various responses in the tumors compared to the standard of care. The primary endpoint is the proportion of patients who achieve a complete cell cycle arrest, defined as a reduction in the proportion of cells staining positive for Ki67, a widely used marker of cancer cell proliferation, as assessed by immunohistochemistry.
64
The Ottawa Hospital will conduct subject enrollment and treatment and evaluate clinical responses, OICR will analyze subject immune responses and conduct biomarker analyses and Ozmosis will manage the data and study in Canada. Intensity will fund the trial and provide INT230-6 supply. Our agreement with OICR, the Ottawa Hospital Research Institute and Dr. Arnout does not provide for any milestone payments, royalties or other compensation to be paid to any party. The agreement provides that each party will solely own any inventions generated in the clinical trial that relate solely to intellectual property owned by that party. Any party may terminate the agreement upon notification that the trial has completed, any party has committed a material breach of the agreement or upon certain bankruptcy events.
Figure 5: Schema for the INVINCIBLE Study in Ontario, Canada
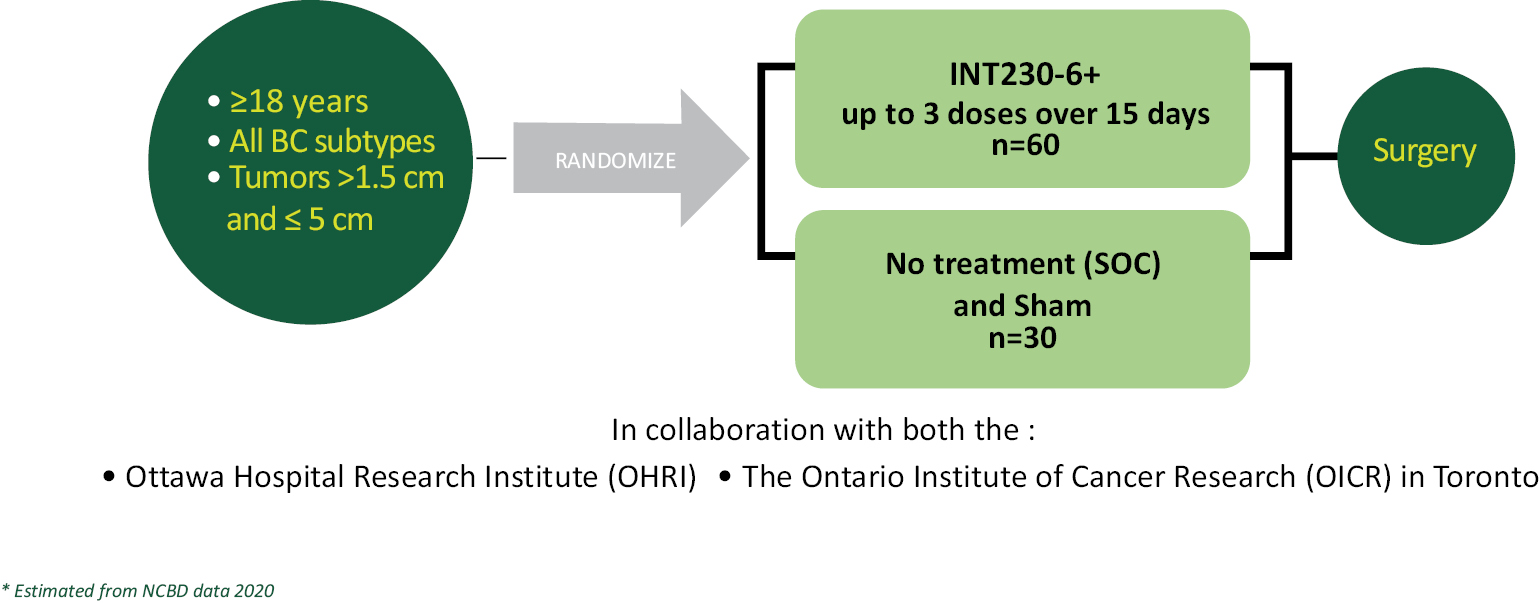
Results from IT-01 Phase 1/2 Clinical Trial
Safety
The Phase 1/2 study has been treating refractory patients, who have failed multiple lines of therapy since May of 2017. Sixty-seven (67) subjects were treated in the Phase 1 escalation portion of study IT-01, which is now complete. The results of the escalation portion indicated a favorable safety profile of INT230-6, with only 8 patients experiencing grade 3 related adverse events in Phase 1. The most frequent related adverse events include localized tumor related pain. The Company has annually submitted safety data from all clinical trials to the FDA and Health Canada. Both regulatory agencies have reviewed the data and have permitted the Company to continue all clinical development programs without comment on safety. The majority of treatment related adverse events have been low grade (grade 1 or 2). As of October 20, 2021, a total of 11 patients out of 95 (12%) have had a grade 3 related adverse event in study IT-01. The grade 3 events have been abdominal pain (4 patients), localized tumor pain (2 patients), fatigue (2 patients), and 1 case each of vomiting, dehydration and dizziness. There have been no grade 4 or 5 treatment related adverse events reported.
Even though our product candidate is dosed directly into the tumor, a key element of safety is to observe how much drug enters the bloodstream. Toxicities are linked to the circulating levels of the active agents in the blood. We measure circulating concentrations of the three main ingredients, SHAO, cisplatin and vinblastine sulfate, in the blood. This type of data is referred to as pharmacokinetics or PK. Data that measured the circulating levels of the key ingredients has been generated from the ongoing study in metastatic patients. The amount of vinblastine sulfate seen in plasma of patients is much lower than a lesser dose given IV. Cisplatin is reduced to metal rapidly and is challenging to measure in blood even for IV dosing. A measurement of vinblastine sulfate provides a better understanding of the PK.
In our study we see free vinblastine plasma concentrations increasing proportional to the amount of drug administered. In essence the concentration of vinblastine seen in the blood increase proportional to the dose given intratumorally. See figure 6 panel A. This effect is independent of the tumor type and highly reproducible. As would be expected the amount of the vinblastine seen in the plasma when given intratumorally is less than 5% of the blood concentrations had the drug been given intravenously. Dosing 80 mL of our drug candidate intratumorally contains 8 mg of vinblastine sulfate and results in ~6.8 nanograms of vinblastine in blood plasma at one hour post dose at 6 hours the amount has dropped to about 2.2 ng/mL. Publications show the plasma concentration of a standard dose of vinblastine sulfate (6.5mg for an averaged sized person) can be estimated to be 72 nanograms per mL (ng/mL) at 1 hour (Owelien J. Cancer Res 8/1977). See figure 6 panel B. We estimate from other studies 240 ng/mL at 6 hours for an IV dose of 5.1 mg (Links, M., Cancer Investigation Volume 17, 1999 -issue 7479-485). The blood plasma concentration profile for of vinblastine at various doses indicates that >95% and perhaps 99% of the drug remains or
65
degrades in the tumor post injection. This drug retention in the tumor spares the patient the side effects of circulating drug. Indeed, the low observed plasma levels of the potent agents following INT230-6 dosing correlates with the low grade of side effects observed. Thus, INT230-6 compares favorably to the toxicities normally associated with cisplatin and vinblastine sulfate when given intravenously at comparable doses.
Figure 6 Free vinblastine (VIN) levels in blood plasma over time for intratumorally (IT) administered INT230-6 compared to IV dosing.
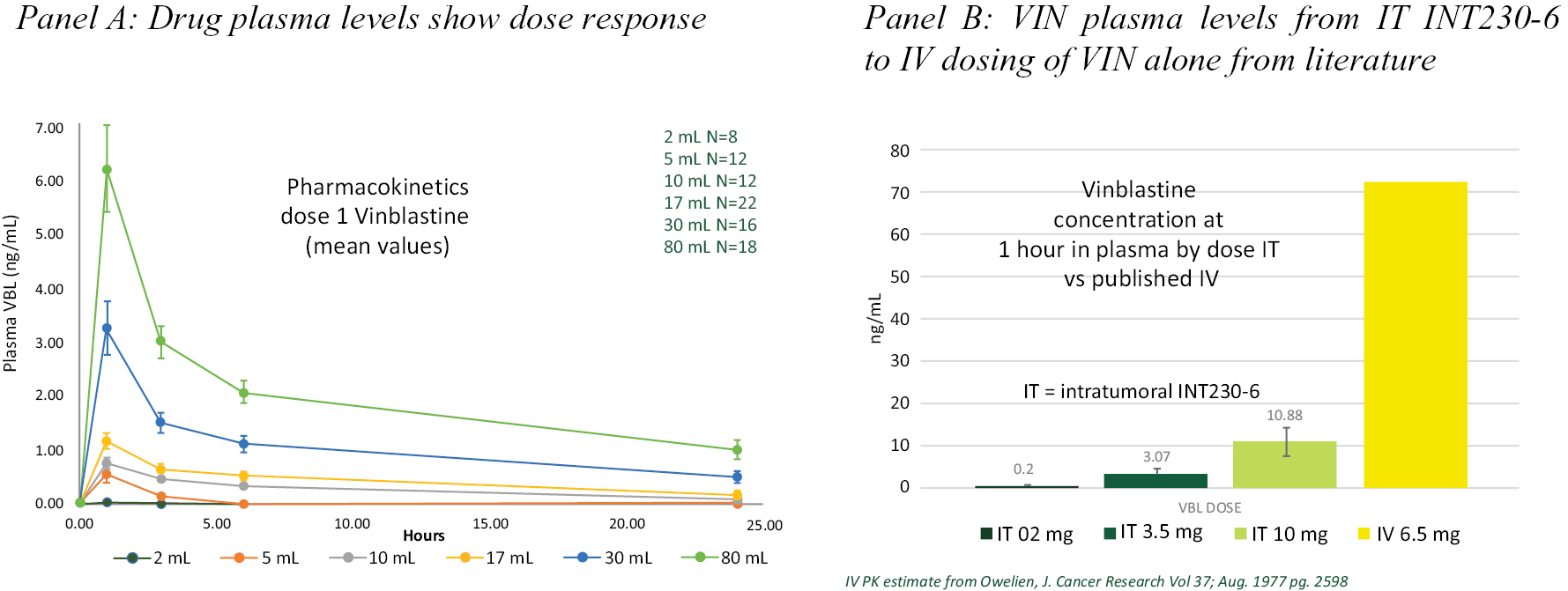
Cytotoxic components in INT230-6 have minimal systemic exposure and short half-life. Most of the active drug remains in the tumor as a result INT230-6 appears to have favorable safety to date.
RECIST
A standard way to measure how well a cancer patient responds to a treatment is based on whether tumors shrink, stay the same, or get bigger. Efficacy assessments for evaluating changes in tumor size in clinical trials are typically conducted with standardized oncology response criteria, for example, Response Evaluation Criteria in Solid Tumors known as RECIST or a newer version 1.1 (RECIST 1.1). There are additional guidelines for immunotherapeutic trials (iRECIST). These criteria measure the change in longest diameter of tumors to assess drug response. An increase in longest diameter of > 20% is considered progressive disease. The rationale behind this is that tumors should generally become smaller. The main benefit of iRECIST is to afford physicians the opportunity to confirm progression with a follow up scan of the tumors 1 to 2 months later. However, both RECIST 1.1 and iRECIST criteria were designed only to assess response to systemic therapies.
Our study initially employed RECIST 1.1, and subsequently, iRECIST methods for determining the efficacy of INT230-6. INT230-6 induced tumor regression in both injected and non-injected lesions in several patients. We have reported data at major medical conferences (ASCO 2021) to indicate that RECIST methodology may not be a good measure of clinical benefit for intratumoral INT230-6.
RECIST 1.1 and iRECIST are not ideal for use with our intratumoral immune based therapy for several reasons. First, the evaluation of injected tumors is complicated by the amount of INT230-6 repeatedly injected and retained in the tumor. Prior to the first efficacy scan, during the first two months (after 5 sessions) of INT230-6 treatment, patients would have received depending on the cohort a dose volume of drug injected into the tumor equivalent to 25% to 250% of the tumor’s volume. As noted above, our data shows a significant percentage of INT230-6 is retained in the tumor, which can increase the tumor size. Second, there is the possibility for immune infiltration. The influx of immune cells also increases tumor size.
Tumor Death (Necrosis)
Investigators report significant necrosis (reduced contrast uptake in the CT image) in many injected tumors including adrenocortical, breast, chordoma, colon, head and neck (H&N), lung, sarcoma and squamous cell. Figure 7 below is an example of a squamous cell tumor that became necrotic by the 2 month scan. The darker contrast of the tumors indicated that significant necrosis of the tumor occurred following treatment.
66
Figure 7 showing that INT230-6 induces tumor necrosis (death) in the injected tumors.
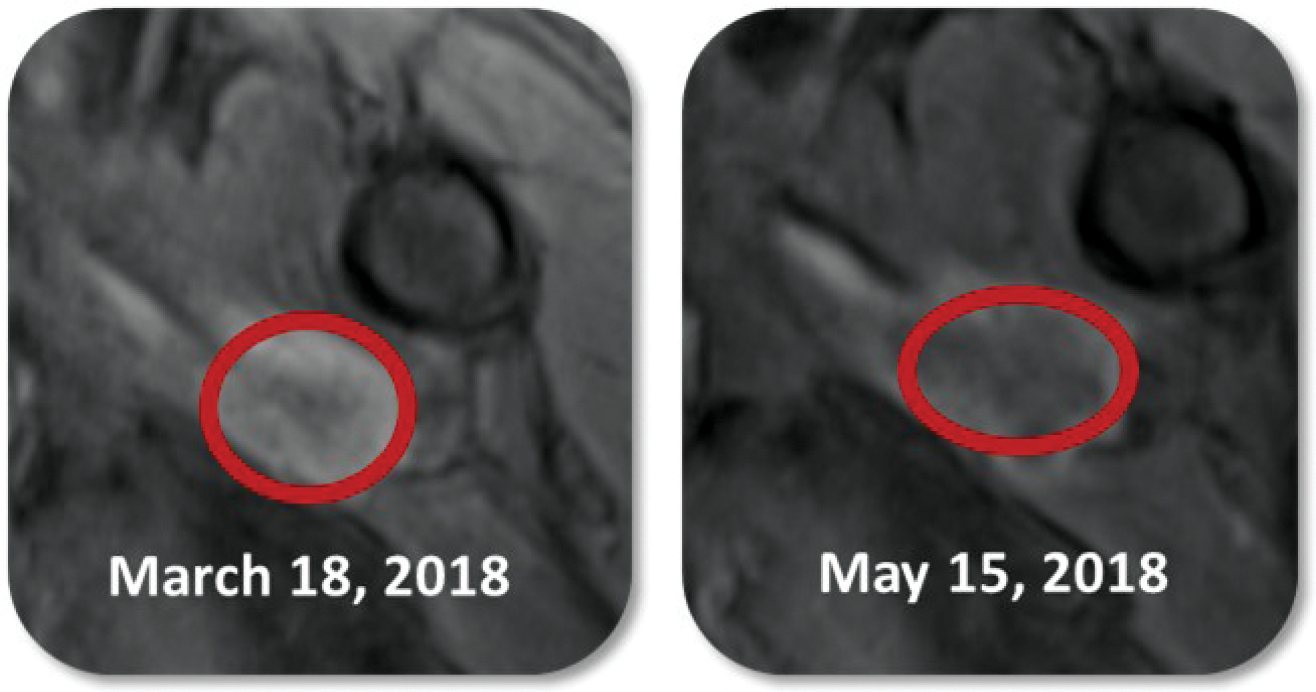
The patient in these images had a squamous cell carcinoma. His cancer continued to progress after 2 surgeries, radiation, and chemotherapy. The patient enrolled in our study in January 2018 with two 10 cm3 deep tumor nodules in his upper arm muscle. The hospital recommended total arm and shoulder amputation. This subject received 4 intratumoral injections equal to 100% of his 2 tumors’ volume. The drug was dosed at ratio of 1 mL per 4 cc of tumor. In the red circle in the left panel there is bright contrast indicating active cancer. At the first scan on May 15, 2018, there was an increase in tumor size, significant necrosis (lack of contrast) and inflammation observed (right panel). The patient received a few doses of Keytruda treatment and had a complete response. This patient has retained his arm and shoulder and is alive as of the last follow up visit in 2021.
Abscopal Effects
In the metastatic study several subjects showed tumor size reduction of non-injected lesions in lymph nodes, liver, lung, perineum, and retroperitoneal areas (i.e. abscopal effects to visceral lesions). The apparent abscopal effect was seen primarily in patients that received a dose greater than 40% of their total tumor burden. Abscopal effect rates may be even higher than known. No tumors under 1 cm in diameter were reported. In addition, many tumors above 1 cm were not followed or reported. We are capturing images from all subjects to be able to determine the true abscopal effect in all subjects at a future time. Figure 8 below shows uninjected tumor diameter changes over time of patients with confirmed reports of abscopal effects.
Figure 8 Change in longest diameter of uninjected tumors over time (abscopal effects).
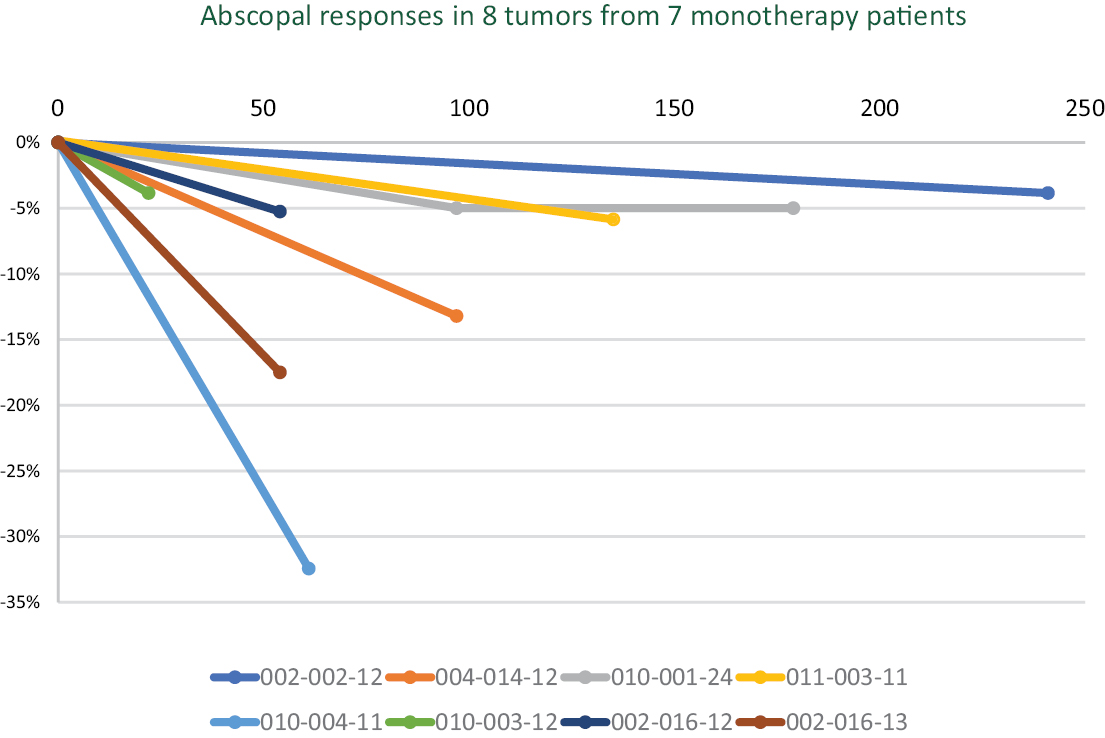
67
Tumor Diameter and Corresponding Volume
For injected tumors, changes in longest diameter often do not correlate with changes in volume. Dosing is completed just prior to their first scan when the increase in tumor diameter is most likely to be highest. As noted above, RECIST measurements of whether a patient’s cancer is stable, decreasing or progressing are based on the changes in the tumor’s longest diameter. An increase in longest diameter above a threshold would indicate progression. In figure 9, the graph on the left shows the change in individual tumors’ longest diameter over time. The graph on the right shows the same tumor’s volume over time. Tumors in many patients treated with INT230-6 can show an increase or no change in longest diameter with a decrease of the corresponding tumor’s volume. There is also a much greater volume decrease than expected for the slight decrease in longest diameter. These data provide further evidence that RECIST may not be a good indication of efficacy for INT230-6.
Figure 9 Using INT230-6 may increase tumor’s longest diameter while decreasing the tumor’s volume.
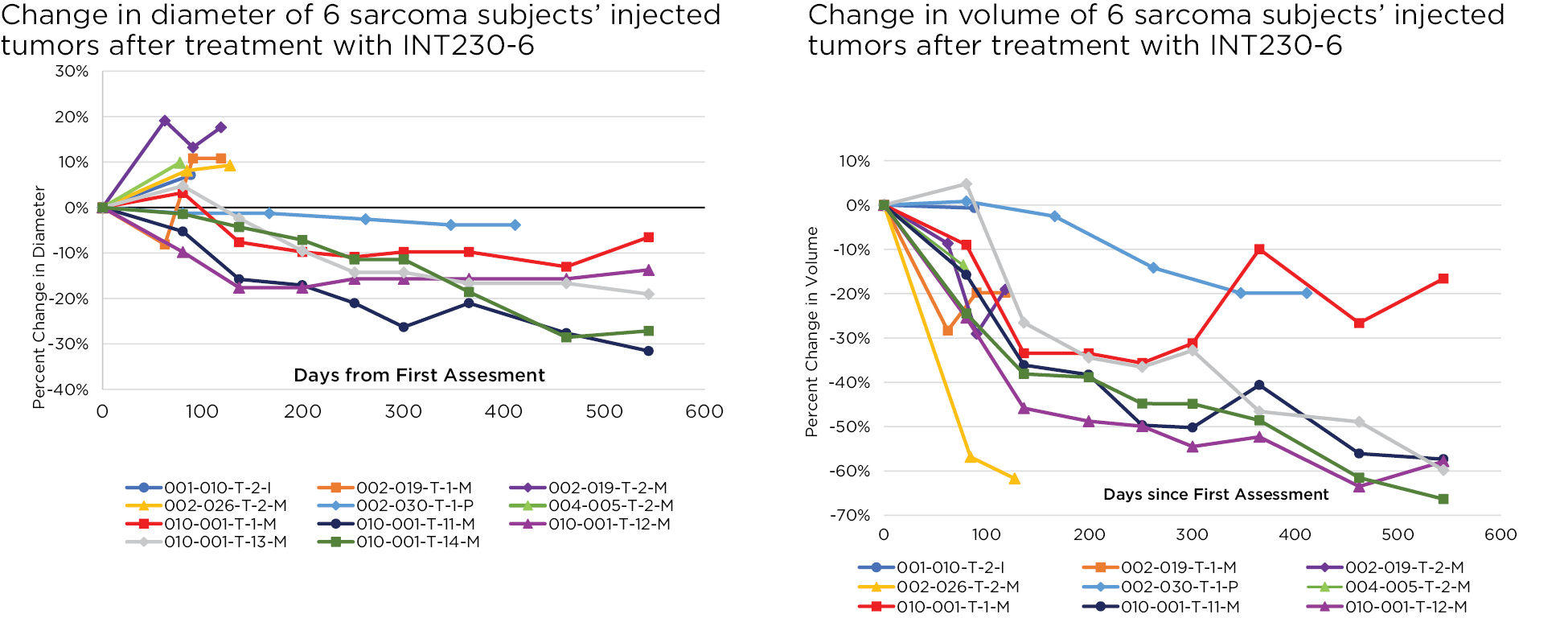
Match the color of each tumor’s line in the left graph (change longest diameter over time) to the same color in the right graph to view that tumor’s change in volume. Data below supports a hypothesis that INT230-6 increases survival in refractory cancer patients. As a result, given the issues with RECIST criteria noted above we believe survival, the FDA’s gold standard efficacy endpoint, is a better measure of our drug candidate’s performance than RECIST methods.
Survival — Phase 1 Basket Studies
The primary objectives of Phase 1 trials are to define the safety or toxicity profile of a new drug and to determine the dose for further evaluation in Phase 2 trials. Patients enrolled in Phase 1 are therefore placed at risk of toxicity, in exchange for an undefined and limited clinical benefit. Furthermore, patients who are considered for Phase 1 trials may be regarded as vulnerable because their physical condition may be deteriorating due to advanced cancer malignancy for which no further standard treatment options exist. Efficacy is not usually the primary objective. Most patients in Phase 1 studies have low survival expectations that ranges from 3 to 8 months depending on the type of cancer and patient’s incoming health. (see Chau, N., BMC Cancer volume 11, Article number: 426 2011).
Over the past two decades the development of a prognostic score to predict survival of patients treated in Phase 1 studies has been completed and validated by the Royal Marsden Hospital (RMH) in the United Kingdom. The score, which ranges from 0 to 3, is highly correlated of overall survival (OS) outcomes. A score of 0 suggests longest potential survival and a 3 worst. Many studies show that subjects enrolled in Phase 1 have survival of under 6 months when RMH scores greater than or equal to 1.
In our study IT-01 patients were enrolled whose cancer progressed following treatment using all approved and some experimental therapies suitable for their specific disease. Forty-three (43%) of patients had previous had an IV form of a platinum-based drug including cisplatin. Forty-four percent (44%) had previously received an anti-PD-1 antibody. Efficacy data from 53 patients enrolled in IT-01 through July 31, 2021 is available from patients receiving INT230-6 alone (referred to as monotherapy). Efficacy data from 16 patients receiving INT230-6 in combination with Keytruda are also available through July 31, 2021. The median number of prior therapies in
68
this population was 4 with a range of 0 to 10 treatments (not including surgery or radiation). As of September 30, 2021, there have been over 689 different deep tumor injections conducted over the course of the trial with over 390 being into visceral deep tumors.
Study IT-01 is a phase 1/2 dose escalation (i.e. the phase 1 basket portion) and phase 2 (expansion of specified cancer types). These types of studies are primarily testing safety in the phase 1 and determining whether there is an efficacy signal in the expansion compared to historical data. There is no control arm in IT-01 and no randomization. Therefore, there is no comparator to determine the significance of any given endpoint. We did observe that patients receiving a dose above a certain percentage of their total tumor burden (more than 40%) had a statistically significantly longer survival than patients who received less than 40%. The subjects receiving a dose >40% of their total incoming tumor burden also lived much longer than would be expected for patients in a phase 1/2 basket study. This indicates a potentially active drug. Given the small size of the population, the heterogeneity of the cancers and variability of the incoming tumor burdens, the high and low dose groups may have been different in a way that we could not measure. We observed a strong signal in just sarcoma patients; however, this population size was also too small to properly assess effectiveness of INT230-6. As a result, we have determined that overall survival, an endpoint that is generally acceptable to FDA for cancer clinical trials, is the most appropriate metric to prove efficacy of our drug candidate. Study IT-01 also indicates that soft tissue sarcoma, at tumor type with high unmet medical need, would be a suitable disease for a large Phase 3 trial.
In our metastatic study survival appears to be impacted by the total dose a patient received relative to number and size of their tumors. Patients receiving a higher percentage of drug (mL) relative to their total tumor burden (cm3) (TTB) remained on study longer regardless of the cancer type. A patient’s total tumor burden is calculated by adding up the volumes of all reported tumors. Simply stated, the more drug given to more tumors, the more likely a subject would be alive longer, though not all tumors need be treated. Killing more of a patient’s cancer is beneficial.
The probability of survival for a given population can be plotted. Figure 10 below illustrates the survival for all monotherapy INT230-6 subjects with total tumor burdens between 2 cm3 and 700 cm3. See Table 1 for the patient population below.
INT230-6 monotherapy population (n=53)
Treating only with our drug candidate 55% of patients would be expected to be alive at one year (gray curve). Subjects dosed an amount of INT230-6 that was less than 40% of their total tumor burden (TTB) had a median overall survival (mOS) between 3 to 6 months. This result is shown in the green curve and is comparable to survival expected in historical Phase 1 basket studies (See Chau, N., BMC Cancer volume 11, Article number: 426 2011). Patients that received a dose of INT230-6 to greater than 40% of their total tumor burden had a ~67% chance of being alive at 1 year. The Hazard Ratio of the blue to green curves was 0.104 with a Confidence Interval (0.04, 0.29) and log rank p=0.000013. These results indicate that survival improves for those dosed to >40% of their tumor burden compared to those receiving under 40%. There were no differences statistically in the two populations with regards to incoming tumor burden; however, the sample size is small. More data from larger randomized controlled in a specific cancer population is required to understand efficacy.
69
Figure 10 Estimates of Survival Dosing INT230-6 alone as of July 31, 2021
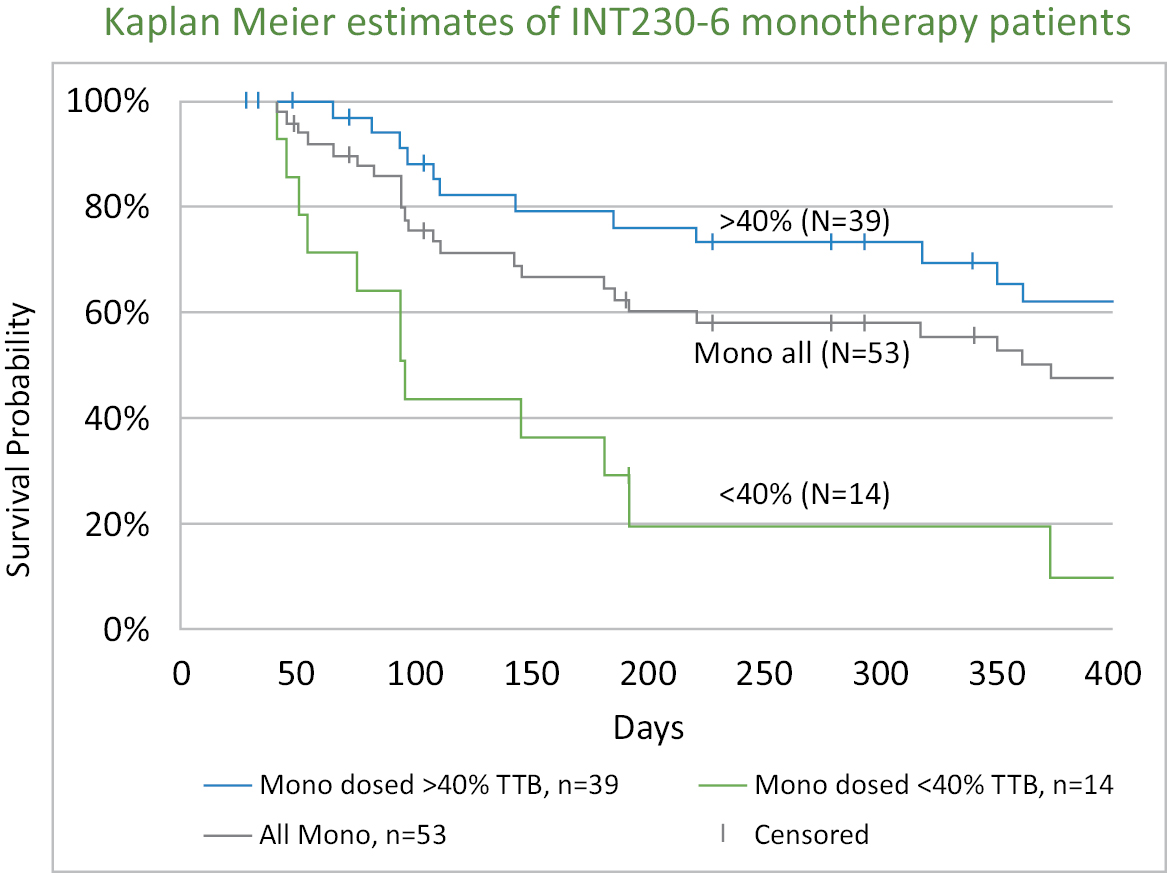
____________
* Monotherapy subjects with reported total tumor burdens >2 cc and <700 cc.
Exploratory analysis of dose relative to total tumor burden (TTB or TB) was conducted. Many tumors, including all under 1 cm in diameter, were not reported and so total tumor burden is likely underestimated.
A preliminary estimate of survival of the combination of INT230-6 and Keytruda tested in a different cancer patient population (see table below) indicates a survival probability of 55% at one year. The combination data also compare favorably to history data from basket studies of patients with these types of refractory cancers; however, the data is still immature and the sample size small at the time of the data cut-off.
Patient Population
INT230-6 with Keytruda (n=16)
Biomarker Analysis
A cancer cell’s surface expresses a unique set of proteins specific to the patient and their cancer type. Certain immune cells can “read” the cell surface to create a patient-specific immune response. However, as noted above, live cancer cells can send signals that can block the immune cells from entering the tumor. There is a constant “cat and mouse” battle between the cancer cell and the immune system.
70
Other local treatments such as radiation or ablation destroy the cell surface. Our technology disperses potent killing agents throughout tumors and enables the potent killing agents to diffuse into the cancer cell without damage to the cell membrane. When the tumor’s cancer cells are no longer alive, the ability of the immune system to identify the cancer and mount a response can be increased.
We collect tumor tissue before and after dosing of our drug candidate from patients injected tumors. We analyze for live and dead cancer cells (referred to as necrotic cells). Our data shows that our drug candidate can kill cancer cells quickly and activate an immune response. We have observed these effects in multiple cancers.
Methods used
Pre-clinical experiments showed that to kill substantial amounts of the tumor would require a dose into the tumor of at least 1 mL of our drug candidate per 4 cubic centimeters of tumor volume with 1 mL for 2 or 3 cc being preferred. INT230-6 injections were conducted on the first treatment cycle’s first day (referred to as C1D0) and on the fourteenth day (C1D14). Pre and post-dose biopsies from the same injected tumor were obtained on C1D0 and again 28 days later just prior to the 3rd dose on the first day of the second treatment cycle (C2D0). To determine the percentage of viable tumor cells and necrotic (dead) cancer cells pre and post two treatments, we conducted analysis on the collected tissue following haemotoxylin and eosin (H&E) staining. H&E tissue analysis helps identify different types of cells and provides important information about the pattern, shape, and structure of cells in a tissue sample.
For many patients, we observed substantial reductions of cancer following the two injections of INT230-6 alone. Below are data on cell killing and immune activation from the two cancer types, breast cancer and sarcoma, for which we are planning Phase 3 programs. We also use immunohistochemistry (IHC) staining to help assess cancer and various immune cell populations, as well as the degree of cancer cell proliferation in the treated tumors.
Results from Breast Cancer Tissue
Several patients with breast cancer have been enrolled throughout the metastatic study. In figure 11, which shows tissue taken from a breast cancer patient (002-022) below, the pre-dose samples stained positive (dark purple) for significant amounts of cancer throughout the sample. However, 28 days later, there was almost no cancer observed in the collected tissue. Magnification is 400µ.
Figure 11 Images from match pair biopsied tissue samples pre and post two INT230-6 injections:

Markers of Cancer Cell Proliferation
There are different rates at which cancer can grow. Highly aggressive cancers have high levels of certain proliferation markers. One such marker is Ki67, a protein found on highly proliferative cells. From the same collected biopsy samples staining was conducted to assess Ki67 in each sample using a validated commercial in vitro diagnostic (IVD) that identifies proliferating cells in all active phases of the cancer’s cell cycle. Both a manual assessment by a pathologist and an automatic image analyzer were used to report values. Results of Ki-67 assessments are shown in the table below.
71
Summary of H&E, tissue analysis and injected tumor volumes in matched pair biopsy samples from 3 triple negative metastatic breast cancer subjects from study IT-01
|
Subject |
% Change in H&E |
Ki67 Values |
%Change in |
%Change in |
Injected tumor |
|||||
|
001-008 |
-86% |
72%/5% |
-93% |
-97% |
4.91 (1.62) |
|||||
|
001-009 |
-50% |
78%/33% |
-58% |
-64% |
46.83 (15.45) |
|||||
|
002-022 |
-72% |
98%/93% |
-5% |
-5% |
51.71 (17.2) |
|||||
|
Average change |
-69% |
-52% |
-55% |
In addition to the substantial reduction of viable cancer cells observed, Ki 67 was reduced significantly. This means the residual cancer is replicating more slowly.
While there are several publications on Ki67 as a predictor of clinical benefit in presurgical settings, the reduction of proliferation markers such as Ki67 is not yet an FDA-recognized approvable endpoint for a drug. However, the percentage of change in pathological complete response (pCR or the absence of cancer in a tumor prior to resection) has been recognized by FDA as a surrogate endpoint to support accelerated approval for neoadjuvant treatment of high-risk, early-stage breast cancer. The INVINCIBLE study will help us to determine whether the addition of INT230-6 to an existing or modified neoadjuvant (presurgical) treatment regimen may increase pCR and reduce toxicities in neoadjuvant triple negative breast cancer subjects.
Immune Response in Breast Cancer Tissue
In preclinical studies, we have shown that our technology reduces the tumor and causes the influx of immune cells (in theory by creating antigen from the dying or dead cancer cells). The below images from a breast cancer patient confirm that this effect also occurs in humans. Applying a special set of stains to the biopsied tissue enables the measurement of immune cells inside the tumor. We observe infiltrating immune cells in the tumor. In figure 12 (below) the first panel (Image A) there is extensive cancer as the blue color seen is for 4´,6-diamidino-2-phenylindole, a blue-fluorescent DNA stain (DAPI) and the marker of live and proliferating cancer. The green and yellow colored cells are of immune cells. The second panel (Image B) shows that at 28 days after the first dose there is a markedly reduced amount of live cancer (no blue stain) indicating significant cancer cell death has occurred. These results are consistent with the H&E results shown above. In addition the green/yellow stained cells, representing CD4 and CD8 anti-cancer T-cells, are increased dramatically throughout the entire tissue. The dead cancer can no longer prevent the immune cells from infiltrating the entire tissue.
Figure 12 IHC Staining of breast cancer tissue for immune cell infiltration pre and post dosing of INT230-6
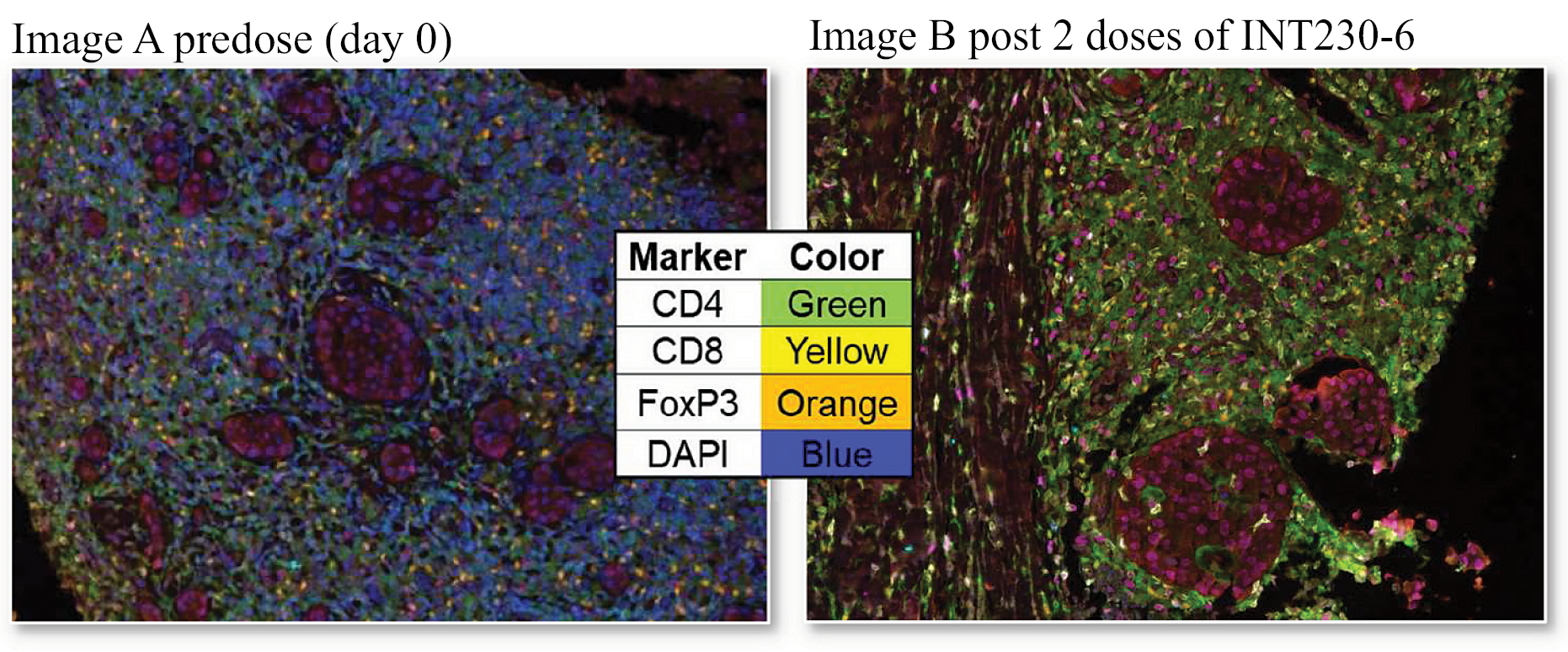
72
Results from Sarcoma Tissue
Several patients with metastatic soft tissue sarcomas have been enrolled in the study. As was seen with breast cancer and multiple other tumor types, there were substantial reductions of cancer in sarcoma patients. An example of this type of result is shown in figure 13. Image A is the stained tissue sample (pre-dose) that shows significant cancer (dark purple cells) throughout the tissue sample. Image B is the stained tissue sample taken on day 28 after two doses of INT230-6 (day 0 and day 14) that shows significant reduction in the cancer (Magnification 3.7x).
Figure 13 Images from match pair biopsied soft tissue sarcoma subject 010-001 pre and post two INT230-6 injections
|
Image A |
Image B |
To confirm the H&E results, we measured DAPI and activated T-cells from a sarcoma tumor. The results again confirm that for this tumor type this is also a substantial reduction of tumor cells as shown by the decrease in the blue DAPI marker post INT230-6 treatment. As was seen in other tumor types, figure 14 show the influx for sarcoma patients into the tumor of CD4 and CD8 T-cells at 28 following the first dose as seen in Images A and B.
Figure 14 Staining of biopsied sarcoma tumor tissue pre and post dosing of INT230-6
|
Image A predose (only DAPI, CD4, CD8) |
Image B post 2 doses (only DAPI, CD4, CD8) |
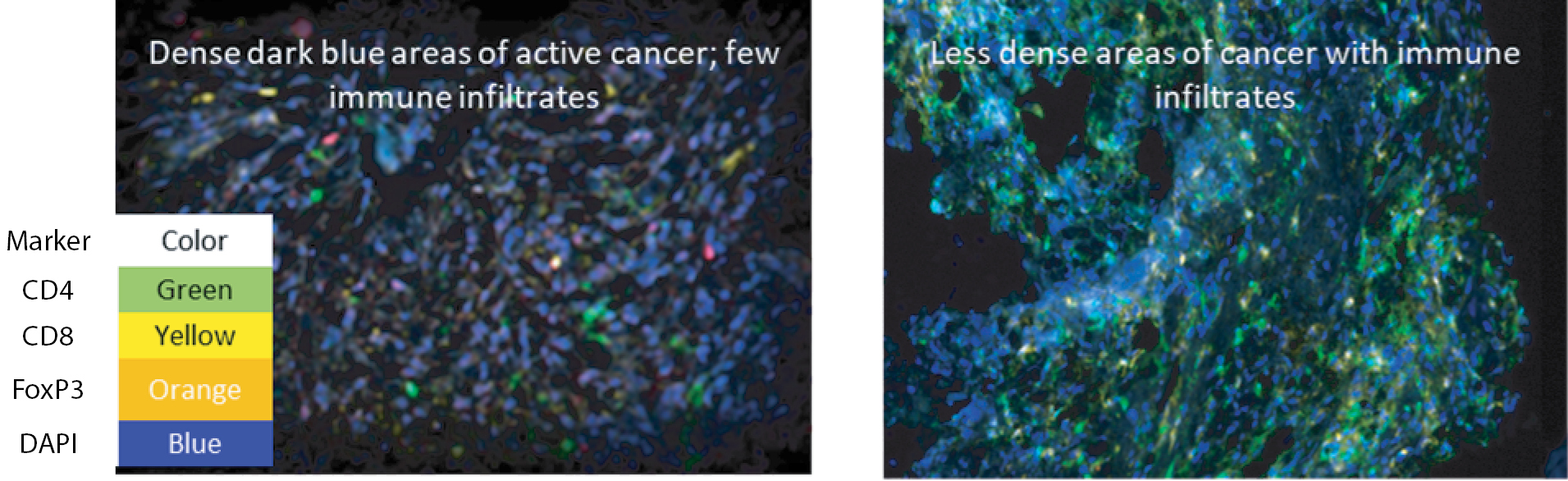
Results of the H&E analysis as well as the multiplex IHC staining show substantial cancer cell reduction, decreases in proliferation, and increased immune infiltration after INT230-6 dosing. The totality of the date indicate the drug has the ability to kill cancer and increase the immune response in multiple cancer types.
73
INT230-6 Efficacy in Soft Tissue Sarcoma
Sarcomas are a rare and heterogeneous group of solid tumors derived from mesenchymal origin. Although single agent or combination anthracycline-based chemotherapy provides some benefit for the treatment of advanced sarcomas, prognosis is still unfavorable with median overall survival of 12 – 16 months and there is significant unmet medical need. By the time subjects fail approved therapies and enter Phase 1 studies patients’ median overall survival is typically 3 – 8 months (see Subbiah, V Scientific Reports | 6:35448 | DOI: 10.1038/srep35448) depending on certain risk factors such those found in the RMHI score.
As of July 31, 2021, 19 patients with sarcoma were treated in study IT-01. Enrolled subjects had a median of 4 (2, 10) prior therapies, median age of 65 and 12% were ECOG 0, 82% ECOG 2 and 6% ECOG 3. The distribution of sarcoma types was 4 Leiomyosarcoma, 3 Liposarcoma, 3 pleomorphic sarcomas, 3 chondrosarcoma, and 2 spindle cell sarcoma, with 1 each of osteosarcoma, myofibroblastic sarcoma, desmoid type, and Kaposi sarcoma. The INT230-6 dose delivered was up to 175 mL (87.5 mg of cisplatin, 17.5 mg of vinblastine sulfate) into one or more tumors at a single visit. The VIN given exceeded the typical 5.1 mg starting IV dose for an average size person. The CIS given was equivalent to a typical IV dose. Safety in sarcoma population remained favorable. The most common treatment-emergent adverse events (TEAEs) in evaluable monotherapy subjects were localized pain, fatigue, decreased appetite, nausea, most of which were low grade.
Survival of subjects in study IT-01 of the refractory population has been favorable as seen in figure 15 below, which shows a probability of survival estimates for the overall population and subgroups. INT230-6 alone or in combination with checkpoint inhibitors appears to show better than expected survival in this refractory population. The Kaplan-Meier (KM) method is used to analyze or estimate “time-to-event” data. The curves used in Kaplan Meier analysis for cancer trials primarily show the estimated time to death by any cause for a given population. For all sarcoma subjects enrolled in our IT-01 study, the Kaplan Meier estimates that ~50% of subjects will be alive at 1 year. Blue curve (n=19). For the population dosed to >40% of their total tumor burden (TTB) the Kaplan Meier estimates ~60% of subjects will be alive at 1 year. Orange curve (n=15). For subjects receiving INT230-6 monotherapy dosed to >40% of their TTB the Kaplan Meier estimates ~60% of subjects will be alive at 1 year. Yellow curve (n=7). Finally, for subjects receiving INT230-6 with a checkpoint drug (88% of which was Yervoy) the Kaplan Meier estimates ~80% of subjects will be alive at 1 year. Purple curve (n=8). Note Data for INT230-6 with Checkpoints is immature as many subjects were recently enrolled.
Figure 15 Estimates of sarcoma subject survival using INT230-6 based on dose per total tumor burden (TTB) from study IT-01
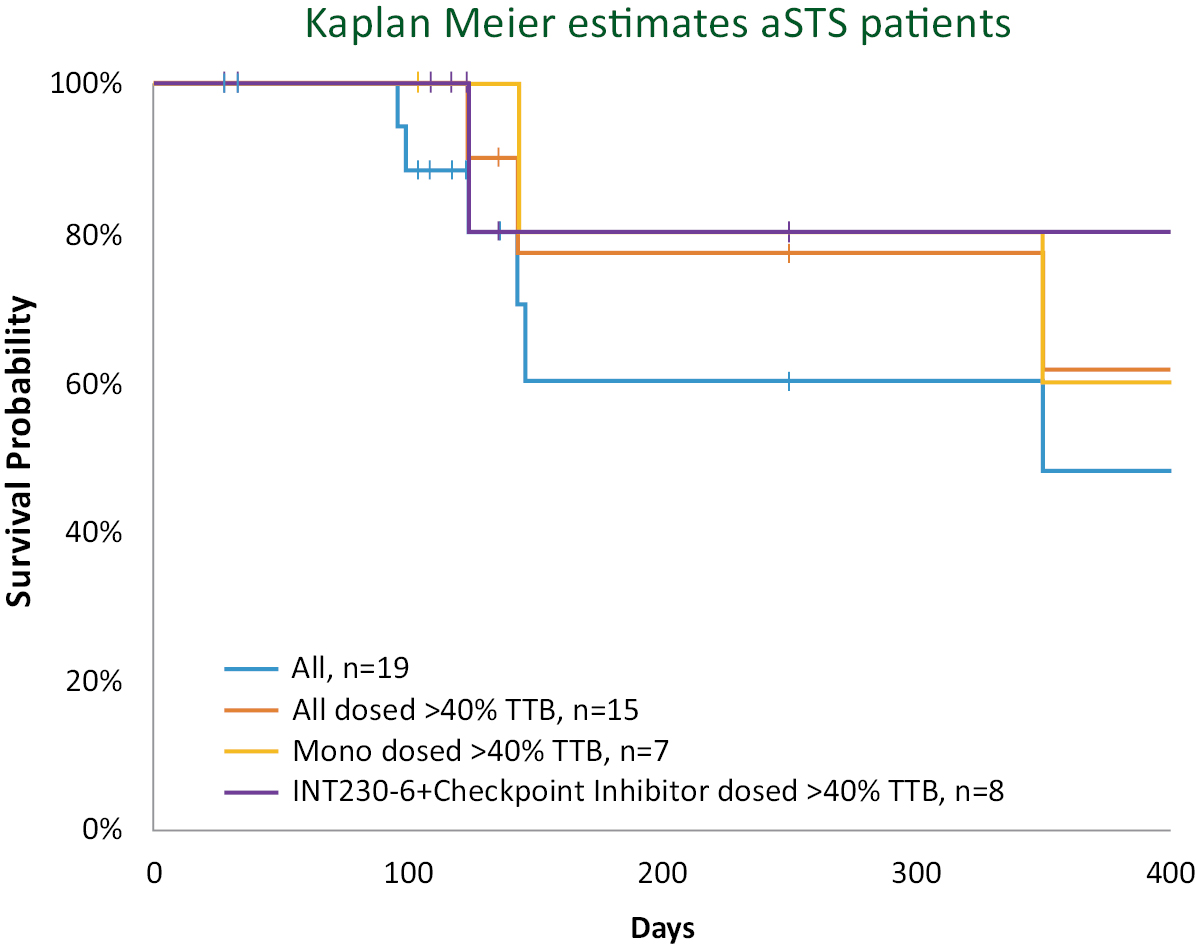
74
Efficacy Results from IT-02 (The INVINCIBLE Study)
Tissue taken via biopsy from tumor in our metastatic study IT-01 shows that viable cancer cells are significantly reduced. However, in our INVINCIBLE study, surgeons also removed the entire breast cancer tumor following INT 230-6 injection. In the INVINCIBLE study when dosed at a ratio proportionate to the tumor’s volume, we showed that greater than 95% of an entire tumor can be killed on a single dose. This result is seen in figure 16 panel A. An ER+PR+HER2+ 3.9 cm grade 3 invasive ductal breast cancer tumor was treated on day 1 with 7.4 cc of INT230-6. Seven days later with another 14.8 cc. The tumor was then resected another seven days later. In panel B, a ER+PR+Her2- 4.4 cm diameter invasive lobular breast cancer tumor was treated with one dose of 21.3 mL of INT230-6, then resected 20 days later. The INT230-6 was able to kill 85% of the ductal tumor. However, in the second panel, the drug was able to diffuse throughout the entire tumor. The boundary of the tumor is shown by the black dotted lines and the red dotted lines show the extent of the necrosis. Pathology conducted on the excised tumor showed that there was only a small percentage of viable cancer cells in one area of the 4.4 cm tumor after a single dose of INT230-6 of 21. 4 mL. More than 95% of the tumor was necrotic (dead) or ghost cells (cells without nucleus). These images show that diffusion distance is proportional to the amount given on a single dose.
Through September 30, 2021, we have treated 20 patients, and we have 10 additional untreated patients in this study. The study plans to enroll a total population of 90 subjects in two parts. The first part will contain 30 patients and evaluates safety and dose. Enrollment for this part is complete. The second part is an expansion that plans to contain an additional 60 patients and will compare tissue taken from a biopsy at the time of diagnosis with tissue taken after surgery on the entire tumor. We will evaluate: (A) the percentage of residual cancer as compared to subjects that receive no treatment or a saline injection, (part 1 showed a significant reduction of cancer in several patients) (B) the change complete cell cycle arrest as measured by Ki67 - a proliferation marker (part 2 only), (C) Surgical outcomes (part 1 showed no delay to surgery), (D) immune response including T cell repertoire (analysis ongoing from part 1), changes in pathology, e.g. necrosis levels, and (E) patient reported outcomes.
As of September 30, 2021, enrollment has been rapid. Patients are highly interested in a product that can potentially destroy the majority of their tumor rapidly while waiting for their surgery and with the possibility to induce an anti-cancer immune response. Surgery has not been delayed or made more difficult by the INT230-6 IT treatment. Adverse events are minimal — mainly transient, low-grade pain at the injection site. Large amounts of necrosis can be induced across multiple breast cancer subtypes with 1 or 2 doses.
The INVINCIBLE study will provide data to help the design of future pivotal studies in the neoadjuvant or pre-surgical settings. In addition, INVINCIBLE results should help to set dosing regimens based on tumor diameter in our Phase 3 metastatic studies.
Figure 16 Showing the extent of the entire tumor and the area of dead cancer for various doses of drug; greater than 95% of the total tumor volume was killed by a single 21 cc injection of INT230-6.
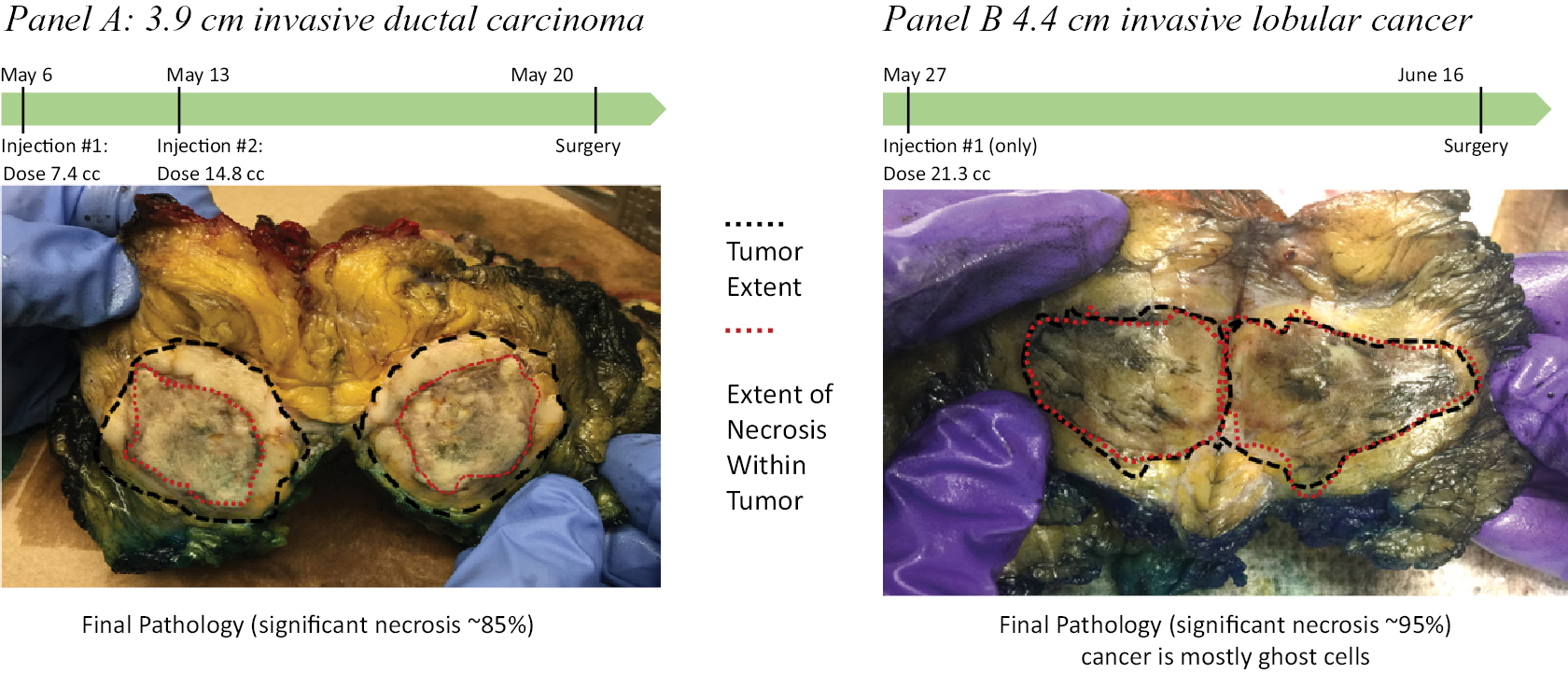
In the above figure the black dotted line shows the extent of the tumor, and red dotted line shows the extent of the necrotic (dead cancer). For a given tumor diffusion distance and thus tumor killing is proportional to the amount of drug dosed. Both tumors shown with high grade (3) proliferative tumors.
75
Planned Phase 3 Trials
Metastatic Sarcoma
Given the positive data on survival seen in our metastatic study in sarcoma patients, we plan to conduct a single Phase 3 study in 2nd/3rd line treatment for locally advanced, recurrent, inoperable, or metastatic non-diffuse soft tissue sarcoma with overall survival as the primary endpoint. The current Phase 3 study design plans to enroll subjects who will be randomized 2 to 1 to either INT230-6 for 5 doses Q2 weeks with maintenance dosing every 9 weeks for 2 years or the standard of care. The three drugs most used for soft tissue sarcoma will be the control SOC at the investigator’s choice depending on the type of sarcoma. Our Phase 3 study is designed to be 90% powered to detect a difference Hazard Value of 0.65 in overall survival between the INT230-6 treatment group and the control group with 331 patients enrolled (2:1 randomization to either INT230-6 treatment or control therapy). The study will have 2 interim data reviews to determine efficacy. The first interim analysis is planned when 50% of the required events (deaths) for the final analysis has occurred and the second analysis will be at 75%. Futility will also be tested as part of the interim analysis. A protocol synopsis was developed and submitted to the FDA. On October 14, 2021, we met with FDA to discuss the Phase 3 protocol and reached alignment on the Phase 3 study design, patient population and statistical approach.
Figure 17 shows the survival curves from five recent Phase 3 studies using now approved standard of care drugs for sarcoma. The figure also shows the expected Phase 3 survival for 1) the blended control based on the likely mix of sarcoma types (green curve) and 2) the expected INT230-6 Phase 3 survival curve that was generated based on our clinical results in sarcoma (navy blue curve). The references showing the Phase 3 data for the standard of care controls are; for trabectadin: Patel S, et. Cancer. 2019 Aug 1;125(15):2610-2620; for eribulin: Schöffski et. al. Lancet. 2016 Apr 16;387(10028):1629-37; and for pazopanib: van der Graaf et. al. Lancet. 2012 May 19;379(9829):1879-86.
It is notable that despite different regimens and sarcoma subtype distributions, the overall survival is consistent for the current standard of care drugs. Our Phase 2 program enrolled sarcoma patients with mixed subtypes whose cancer progressed despite a median of 3 prior treatments. We plan to enroll a similar mix of sarcoma patients; in Phase 3, however, no patient will have progressed on more than 2 treatments. Thus, patients in our planned Phase 3 study will be healthier than those treated in our Phase 2 study. Over 25% of patients in our Phase 2 study were underdosed. In the planned Phase 3 study such a high percentage of underdosed patient is unlikely. Patients in the planned Phase 3 program will also receive long term maintenance treatment of INT230-6 every 9 weeks, which mostly did not occur in our Phase 2 program.
Figure 17 the expected Phase 3 INT230-6 survival curve compared to the approved standard of care drugs
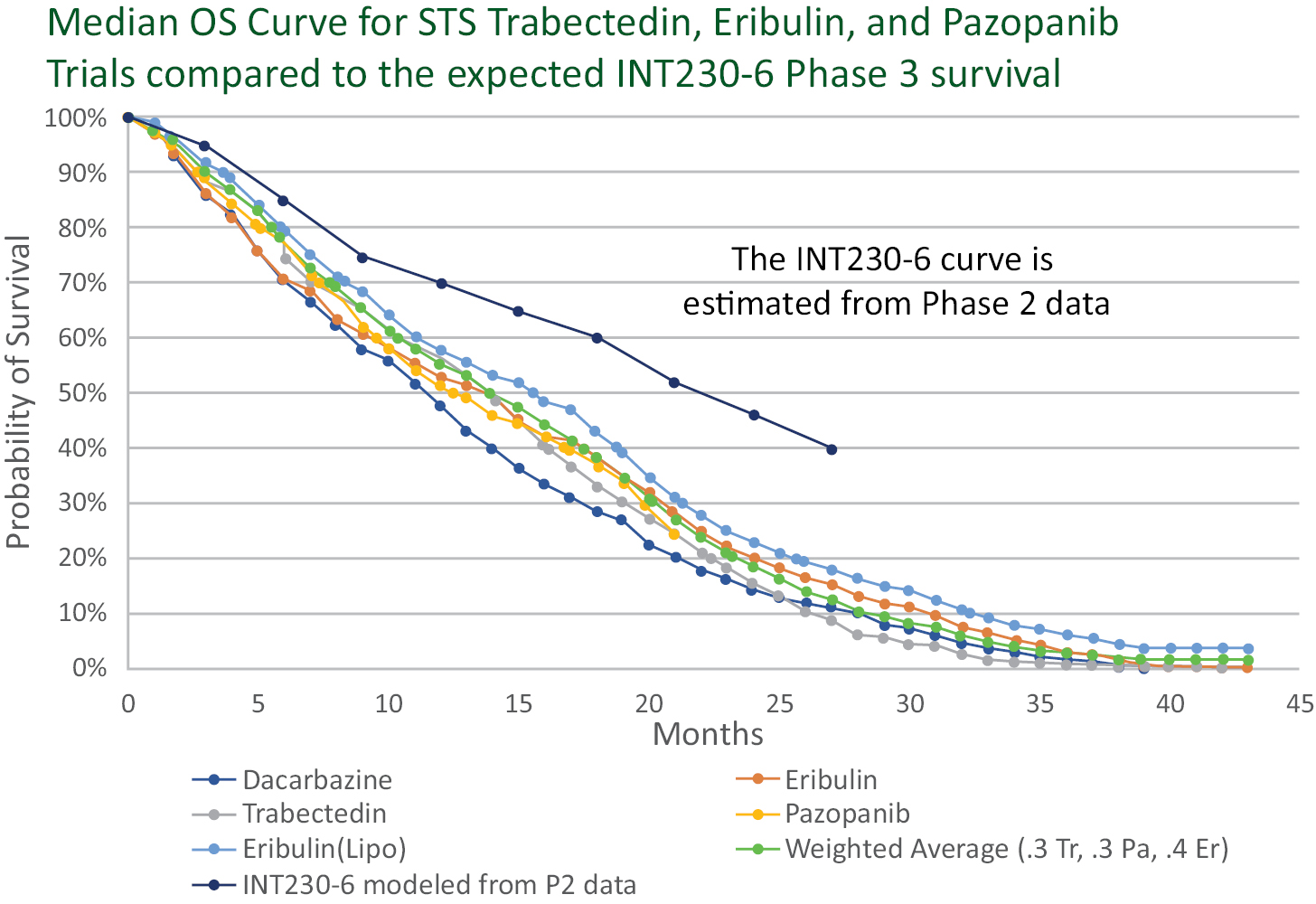
76
The current Phase 3 design proposes an endpoint of overall survival in all advanced soft tissue sarcoma patients. In addition, we proposed to have 2 interim analyses; the first at 50% and the second at 75% of expected events needed for the final analysis. We plan to enroll 2 subjects in the INT230-6 group per one subject of any of the three used drugs for each patient’s type of advanced soft tissue sarcoma. INT230-6 will be dosed every 2 weeks for 5 doses with maintenance every 9 weeks. The SOC drugs will be dosed at their approved regimens.
Metastatic Triple Negative Breast Cancer
The FDA designation of INT230-6 for Fast Track was made in response to our proposed development program evaluating INT230-6 for the treatment of patients with relapsed or metastatic triple negative breast cancer.
• In 2021, over 270,000 patients were expected to be diagnosed with breast cancer;
• Of these, ~11-17% of tumors found in breast cancer patients will not test positive for any of human epidermal growth factor receptor 2 (HER2), estrogen receptor (ER), or progesterone receptor (PR) proteins, and thus will be classified as triple negative tumors;
• TNBC is commonly found in younger patients (<50), African American and Hispanic women, and patients with a BRCA1 mutation (~70% of BRCA patients are triple negative); and
• TNBC tends to be more aggressive, i.e., higher grade, relative to other forms of breast cancer with limited treatment options, highlighting a high degree of unmet need in this patient population.
TNBC patients have a poor prognosis, with a median overall survival of 13.3 months with treatment first line. Recently approved treatments including Lynparza (PARP inhibitor) and Tecentriq (PD-L1 inhibitor). Those treatments target a specific subset of patients, with BRCA1/2 and PD-L1 positive markers, respectively. Our target population would be more inclusive.
Continuing chemotherapy treatment until disease progression is currently the standard of care for patients with metastatic TNBC, with no preferred chemotherapy regimens established at this time. Gilead presented data at the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting (Abstract #1080) for second line use of sacituzumab (Trodelvy). Sacituzumab extended median overall survival to 10.9 months versus 4.9 months with chemotherapy (HR: 0.51; 95% CI: 0.28-0.91).
With a small sample size in study IT-01 INT230-6 either as monotherapy or with pembrolizumab has shown in refractory metastatic breast cancer (all types) a median overall survival of 12 months (n=9), and in subset of just m TNBC subjects, a median overall survival of approximately 12.5 months.
INT230-6 Phase 2/3 study design would consist of metastatic TNBC patients whose cancer has progressed following 1 to 2 lines of prior therapy. The Phase 2 study would be approximately 60 patients with INT230-6 arm and a control arm cohort design of patients using investigators choice of therapy. The endpoints would be median overall survival. Patients would receive 5 doses of INT230-6 every two weeks delivered IT with a maintenance dosing. The protocol will be designed to allow us to determine, within 12 months following completion of enrolment, whether INT230-6 has the potential to offer clinical benefit. A combination of INT230-6 with a checkpoint antibody (e.g. pembrolizumab or ipilimumab) within the randomized Phase 2 may be considered. From the results of the ongoing Phase 2, the company would make a strategic decision to use either monotherapy or combination with a checkpoint and size the final study accordingly. Phase 3 would be randomized 2 to 1 against investigators choice of treatment. Additional subjects could be added to the Phase 2 portion to complete the Phase 3 program.
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of drugs such as those we are developing. We, along with our vendors, collaboration partners, CROs and contract manufacturers, will be required to navigate the various preclinical, clinical, manufacturing and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our product candidate. The process of obtaining regulatory approvals of drugs and ensuring subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources.
77
In the United States, where we are initially focusing our product development, the FDA regulates drugs under the federal Food, Drug, and Cosmetic Act (FDCA) and its implementing regulations. Drug products are also subject to other federal, state and local statutes and regulations. Our product candidate is early-stage and has not been approved by the FDA for marketing in the United States.
The process required by the FDA before our product candidate is approved for therapeutic indications and may be marketed in the United States generally involves the following:
• completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with Good Laboratory Practice, or GLP, requirements;
• submission to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made;
• approval by an Institutional Review Board, or IRB, or independent ethics committee at each clinical trial site before each trial may be initiated;
• performance of adequate and well-controlled clinical trials in accordance with Good Clinical Practice, or GCP requirements and other clinical trial-related regulations to establish the safety and efficacy of the proposed drug product candidate for its intended purpose;
• preparation and submission to the FDA of a New Drug Application (NDA) after completion of all pivotal trials;
• a determination by the FDA of its receipt of an NDA, to file the application for review;
• satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the product will be produced to assess compliance with current Good Manufacturing Practice requirements, or CGMPs, to assure that the facilities, methods and controls are adequate to assure the drug product’s identity, strength, quality and purity;
• potential FDA audit of the clinical trial sites that generated the data in support of the NDA;
• payment of user fees for FDA review of the NDA; and
• FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug product in the United States.
Preclinical and clinical trials for drug products
Before testing any drug in humans, the product candidate must undergo rigorous preclinical testing. Preclinical studies include laboratory evaluations of chemistry, formulation and stability, as well as in vitro and animal studies to assess safety and in some cases to establish the rationale for therapeutic use. The conduct of preclinical studies is subject to federal and state regulations and requirements, including GLP requirements for safety and toxicology studies. The results of the preclinical studies, together with manufacturing information and analytical data must be submitted to the FDA as part of an IND. An IND is a request for authorization from the FDA to administer an investigational product to humans, and must become effective before clinical trials may begin. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks, and imposes a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Some long-term preclinical testing may continue after the IND is submitted. Accordingly, submission of an IND may or may not result in FDA authorization to begin a trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development of a product candidate, and the FDA must grant permission, either explicitly or implicitly by not objecting, before each clinical trial can begin.
78
The clinical stage of development involves the administration of the product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirements that all research subjects provide their informed consent for their participation in any clinical trial. For cancer patients, the Phase 1 usually involves patients whose cancer has progressed following all approved therapies for that particular cancer.
Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters and criteria to be used in monitoring safety and evaluating effectiveness. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable related to the anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completed. The FDA, the IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trials to public registries. Information about applicable clinical trials, including clinical trials results, must be submitted within specific timeframes for publication on the www.clinicaltrials.gov website. Disclosure of the results of such trials can be delayed in some cases for up to two years after the date of completion of the trial. Failure to timely register a covered clinical trial or to submit trial results as provided for in the law can give rise to civil monetary penalties and also prevent the non-compliant party from receiving future grant funds from the federal government. The NIH’s Final Rule on ClinicalTrials.gov registration and reporting requirements became effective in 2017, and both NIH and FDA have signaled the government’s willingness to begin enforcing those requirements against non-compliant clinical trial sponsors.
We have conducted our trials in Canada under a Clinical Trial Agreement with Health Canada, the regulatory authority in Canada. While we plan to conduct any international clinical trials under appropriate country filings in the future, a sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. The FDA will accept a well-designed and well-conducted foreign clinical study not conducted under an IND if the study was conducted in accordance with GCP requirements, and the FDA is able to validate the data through an onsite inspection if deemed necessary.
Clinical trials to evaluate therapeutic indications to support NDAs for marketing approval are typically conducted in three sequential phases, which may overlap.
• Phase 1 — Phase 1 clinical trials involve initial introduction of the investigational product into healthy human volunteers or patients with the target disease or condition. These studies are typically designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, evaluate the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. As noted above for new cancer treatments such as ours, the Phase 1 involves patients whose cancer has progressed following all approved therapies for that particular cancer not healthy volunteers.
• Phase 2 — Phase 2 clinical trials typically involve administration of the investigational product to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials.
• Phase 3 — Phase 3 clinical trials typically involve administration of the investigational product to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. Generally, one or two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA.
79
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators fifteen days after the trial sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse events, findings from other studies or animal or in vitro testing that suggest a significant risk for human participants exposed to the investigational product and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must also notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but in no case later than seven calendar days after the sponsor’s initial receipt of the information.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the drug characteristics of the product candidate and finalize a process for manufacturing the drug product in commercial quantities in accordance with CGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and manufacturers must develop, among other things, methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life and to identify appropriate storage conditions for the product candidate.
New Drug Applications (NDA) Submission and Review by the FDA
We intend to seek data exclusivity or market exclusivity for INT230-6. Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. An NDA is a request for approval to market a new drug for one or more specified indications. The NDA must include all relevant data available from pertinent pre-clinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. FDA approval of an NDA must be obtained before a chemical drug may be marketed in the United States.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA must contain data to assess the safety and effectiveness of the drug product candidate for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The Food and Drug Administration Safety and Innovation Act requires that a sponsor who is planning to submit a marketing application for a drug product that includes a new clinically active component, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan (PSP) within sixty days after an end-of-Phase 2 meeting or as may be agreed between the sponsor and FDA. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug product for an indication for which orphan designation has been granted.
The FDA reviews all submitted NDAs before it accepts them for filing, and may request additional information rather than accepting the NDA for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt, and such decision could include a refusal to file by the FDA. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the NDA. The FDA reviews an NDA to determine, among other things, whether the product is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued identity, strength, quality and purity. Under the goals and polices agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA targets ten months, from the filing date, in which to complete its initial review of an original NDA and respond to the applicant, and six months from the filing date of an original NDA filed for priority review. The FDA does not always meet its PDUFA goal dates for standard or priority NDAs, and the review process is often extended by FDA requests for additional information or clarification.
80
Further, under PDUFA, as amended, each NDA must be accompanied by a substantial user fee, and the sponsor of an approved NDA is also subject to an annual program fee for each approved drug product. FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions may be available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA may refer an application for a new drug product to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, which reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP and other requirements and the integrity of the clinical data submitted to the FDA.
The FDA also may require submission of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition for approving the NDA to ensure that the benefits of the product outweigh its risks. The REMS could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk-minimization tools.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter or a Complete Response Letter. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter will usually describe all of the deficiencies that the FDA has identified in the NDA, except that where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the Complete Response Letter without first conducting required inspections, testing submitted product lots, and/or reviewing proposed labeling. In issuing the Complete Response Letter, the FDA may recommend actions that the applicant might take to place the NDA in condition for approval, including requests for additional information or clarification. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications.
Even if the FDA approves a product, depending on the specific risk(s) to be addressed, the FDA may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a product’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Expedited development and review programs for drugs
The FDA maintains several programs intended to facilitate and expedite development and review of new drugs to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These programs include Fast Track Designation, Breakthrough Therapy designation, priority review and Accelerated Approval.
A new drug product is eligible for Fast Track Designation if it is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address unmet medical needs for such disease or condition. Fast Track Designation applies to the combination of the product and the specific indication for which it is being studied. Fast Track Designation provides increased opportunities for sponsor interactions with the FDA during preclinical and clinical development, in addition to the potential for rolling review once a marketing application is filed, meaning that the FDA may consider for
81
review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
In addition, a new drug product may be eligible for Breakthrough Therapy designation if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug, alone or in combination with one or more other drugs or biologics, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough Therapy designation provides all the features of Fast Track Designation in addition to intensive guidance on an efficient development program beginning as early as Phase 1, and FDA organizational commitment to expedited development, including involvement of senior managers and experienced review staff in a cross-disciplinary review, where appropriate.
Any product submitted to the FDA for approval, including a product with Fast Track or Breakthrough Therapy designation, may also be eligible for additional FDA programs intended to expedite the review and approval process, including priority review and Accelerated Approval. A product is eligible for priority review if it is intended to treat a serious or life-threatening disease or condition, and if approved, would provide a significant improvement in safety or effectiveness. For original NDAs, priority review designation means the FDA’s goal is to take action on the marketing application within six months of the 60-day filing date (compared with ten months under standard review).
A product intended to treat serious or life-threatening diseases or conditions may receive Accelerated Approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than on irreversible morbidity or mortality which is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
Accelerated Approval is usually contingent on a sponsor’s agreement to conduct additional post-approval studies to verify and describe the product’s clinical benefit. The FDA may withdraw approval of a drug approved under Accelerated Approval if, for example, the sponsor fails to conduct the confirmatory trials in a timely manner or the confirmatory trial fails to verify the predicted clinical benefit of the product. In addition, unless otherwise informed by the FDA, the FDA currently requires, as a condition for Accelerated Approval, that all advertising and promotional materials that are intended for dissemination or publication within 120 days following marketing approval be submitted to the agency for review during the pre-approval review period, and that after 120 days following marketing approval, all advertising and promotional materials must be submitted at least 30 days prior to the intended time of initial dissemination or publication.
Fast Track Designation, Breakthrough Therapy designation, priority review and Accelerated Approval do not change the scientific or medical standards for approval or the quality of evidence necessary to support approval but may expedite the development or review process.
U.S. post-approval requirements for drugs
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, reporting of adverse experiences with the product, complying with promotion and advertising requirements, which include restrictions on promoting products for unapproved uses or patient populations (known as “off-label use”) and limitations on industry-sponsored scientific and educational activities. Although physicians may prescribe approved products for off-label uses, manufacturers may not market or promote such uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, including not only by our employees but also by agents of us or those speaking on our behalf, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties, including liabilities under the False Claims Act where products carry reimbursement under federal health care programs. Promotional materials for approved drugs must be submitted to the FDA in conjunction with their first use or first publication. Further, if there are any modifications to the product, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require the development of additional data or preclinical studies and clinical trials.
82
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-market testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and their subcontractors involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMP, which impose certain procedural and documentation requirements upon us and our contract manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from CGMP and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. Failure to comply with statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, product seizures, injunctions, civil penalties or criminal prosecution. There is also a continuing, annual program fee for any marketed product.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, requirements for post-market studies or clinical trials to assess new safety risks, or imposition of distribution or other restrictions under a REMS. Other potential consequences include, among other things:
• restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
• safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings or other safety information about the product;
• mandated modification of promotional materials and labeling and issuance of corrective information;
• fines, warning letters, or untitled letters;
• holds on clinical trials;
• refusal of the FDA to approve applications or supplements to approved applications, or suspension or revocation of product approvals;
• product seizure or detention, or refusal to permit the import or export of products;
• injunctions or the imposition of civil or criminal penalties; and
• consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs.
Orphan Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation, or ODD, to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with either a patient population of fewer than 200,000 individuals in the United States, or a patient population greater of than 200,000 individuals in the United States when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States of that drug or biologic. ODD must be requested before submitting an NDA. After the FDA grants ODD, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has received ODD and subsequently receives the first FDA approval for a particular clinically active component for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same biologic for the same indication for seven years from the approval of the NDA, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the
83
orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of ODD are tax credits for certain research and a waiver of the NDA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received ODD. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
A drug product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study. The data from such study do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. Although this is not a patent term extension, it effectively extends the regulatory period during which the FDA cannot approve another application.
The Hatch-Waxman Act and Marketing Exclusivity
Under the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments to the FDCA, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute and also enacted Section 505(b)(2) of the FDCA. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application, or ANDA, to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing conducted for a drug product previously approved under an NDA, known as the reference listed drug (RLD). Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug. In contrast, Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. A Section 505(b)(2) applicant may eliminate the need to conduct certain preclinical or clinical studies, if it can establish that reliance on studies conducted for a previously approved product is scientifically appropriate. Unlike the ANDA pathway used by developers of bioequivalent versions of innovator drugs, which does not allow applicants to submit new clinical data other than bioavailability or bioequivalence data, the 505(b)(2) regulatory pathway does not preclude the possibility that a follow-on applicant would need to conduct additional clinical trials or nonclinical studies; for example, they may be seeking approval to market a previously approved drug for new indications or for a new patient population that would require new clinical data to demonstrate safety or effectiveness. The FDA may then approve the new product for all or some of the label indications for which the RLD has been approved, or for any new indication sought by the Section 505(b)(2) applicant, as applicable.
In seeking approval of an NDA or a supplement thereto, the NDA sponsor is required to list with the FDA each patent with claims that cover the sponsor’s product or an approved method of using the product. Upon approval of an NDA, each of the patents listed in the application for the drug is published in the FDA publication Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. When an ANDA applicant submits its application to the FDA, the applicant is required to certify to the FDA concerning any patents listed in the Orange Book for the RLD, except for patents covering methods of use for which the follow-on applicant is not seeking approval. To the extent a Section 505(b)(2) applicant is relying on studies conducted for an already approved product, such an applicant is also required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would.
Specifically, any applicant who subsequently files an ANDA or 505(b)(2) NDA that references the drug listed in the Orange Book must certify to the FDA that with respect to each published patent, (i) the required patent information has not been filed by the original applicant of the RLD; (ii) the listed patent already has expired; (iii) the listed patent has not expired, but will expire on a specified date and approval is sought after patent expiration; or (iv) the listed patent is invalid, unenforceable or will not be infringed by the manufacture, use or sale of the new product. These are known as Paragraph I, II, III, and IV certifications, respectively.
84
If a Paragraph I or II certification is filed, the FDA may make approval of the application effective immediately upon completion of its review. If a Paragraph III certification is filed, the approval may be made effective on the patent expiration date specified in the application, although a tentative approval may be issued before that time. If an application contains a Paragraph IV certification, a series of events will be triggered, the outcome of which will determine the effective date of approval of the ANDA or 505(b)(2) application.
A certification that the new product will not infringe the RLD’s listed patents or that such patents are invalid is called a Paragraph IV certification. If the follow-on applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders for the RLD once the applicant’s NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a legal challenge to the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of their receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA or 505(b)(2) NDA until the earlier of 30 months after the receipt of the Paragraph IV notice, expiration of the patent or a decision in the infringement case that is favorable to the ANDA or 505(b)(2) applicant. Alternatively, if the listed patent holder does not file a patent infringement lawsuit within the required 45-day period, the follow-on applicant’s ANDA or 505(b)(2) NDA will not be subject to the 30-month stay.
In addition, under the Hatch-Waxman Amendments, the FDA may not approve an ANDA or 505(b)(2) NDA until any applicable period of non-patent exclusivity for the referenced RLD has expired. These market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a drug containing a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement.
The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving follow-on applications for drugs containing the original active agent. Five-year and three-year exclusivity also will not delay the submission or approval of a traditional NDA filed under Section 505(b)(1) of the FDCA. However, an applicant submitting a traditional NDA would be required to either conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five-year patent term extension. The allowable patent term extension is calculated as half of the drug’s testing phase — the time between when the IND becomes effective and NDA submission — and all of the review phase — the time between NDA submission and approval, up to a maximum of five years. The time can be shortened if FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years. For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the Patent and Trademark Office (PTO) must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA has not been submitted.
Other regulatory matters
Manufacturing, sales, promotion and other activities of drug products following approval, where applicable, or commercialization are also subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, which may include the Centers for Medicare & Medicaid Services, or CMS, other divisions of the Department of
85
Health and Human Services, or HHS, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments and governmental agencies.
Other healthcare laws
Healthcare providers, physicians, and third-party payors will play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. Our business operations and any current or future arrangements with third-party payors, healthcare providers and physicians may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we develop, market, sell and distribute any drugs for which we obtain marketing approval. In the United States, these laws include, without limitation, state and federal anti-kickback, false claims, physician transparency, and patient data privacy and security laws and regulations, including but not limited to those described below.
• The federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, paying, receiving or providing any remuneration (including any kickback, bribe, or certain rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; a person or entity need not have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it in order to have committed a violation. Violations are subject to civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs;
• The federal civil and criminal false claims laws, including the civil False Claims Act, or FCA, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment or approval that are false, fictitious or fraudulent; knowingly making, using, or causing to be made or used, a false statement or record material to a false or fraudulent claim or obligation to pay or transmit money or property to the federal government; or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay money to the federal government. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. In addition, the government may assert that a claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. The FCA also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. When an entity is determined to have violated the federal civil False Claims Act, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs;
• The federal civil monetary penalties laws, which impose civil fines for, among other things, the offering or transfer or remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies;
• The Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for knowingly and willfully executing a scheme, or attempting to execute a scheme, to defraud any healthcare benefit program, including private payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, or falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity need not have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
• HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, imposes, among other things, specified requirements on covered entities and their business associates relating to the privacy and security
86
of individually identifiable health information including mandatory contractual terms and required implementation of technical safeguards of such information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates in some cases, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions;
• The Physician Payments Sunshine Act, enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, imposed new annual reporting requirements for certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, for certain payments and “transfers of value” provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. In addition, many states also require reporting of payments or other transfers of value, many of which differ from each other in significant ways, are often not pre-empted, and may have a more prohibitive effect than the Sunshine Act, thus further complicating compliance efforts. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made in the previous year to certain non-physician providers such as physician assistants and nurse practitioners;
• Federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and
• Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party-payors, including private insurers, and may be broader in scope than their federal equivalents; state and foreign laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, and restrict marketing practices or require disclosure of marketing expenditures and pricing information; state and foreign laws that govern the privacy and security of health information in some circumstances. These data privacy and security laws may differ from each other in significant ways and often are not pre-empted by HIPAA, which may complicate compliance efforts.
The distribution of drug products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other related governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, reputational harm, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar settlement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to similar actions, penalties and sanctions. Ensuring business arrangements comply with applicable healthcare laws, as well as responding to possible investigations by government authorities, can be time- and resource-consuming and can divert a company’s attention from its business.
87
Coverage and Reimbursement
In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Thus, even if a product candidate is approved, sales of the product will depend, in part, on the extent to which third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercial health insurers and managed care organizations, provide coverage, and establish adequate reimbursement levels for, the product. In the United States, no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors are increasingly challenging the prices charged, examining the medical necessity, and reviewing the cost-effectiveness of medical products and services and imposing controls to manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. Additionally, companies may also need to provide discounts to purchasers, private health plans or government healthcare programs. Nonetheless, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover a product could reduce physician utilization once the product is approved and have a material adverse effect on sales, our operations and financial condition. Additionally, a third-party payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of products have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’s revenue generated from the sale of any approved products. Coverage policies and third-party payor reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which a company or its collaborators receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Healthcare reform
In the United States and some foreign jurisdictions, there have been, and likely will continue to be, a number of legislative and regulatory changes and proposed changes regarding the healthcare system directed at broadening the availability of healthcare, improving the quality of healthcare, and containing or lowering the cost of healthcare. For example, in March 2010, the United States Congress enacted the ACA, which, among other things, includes changes to the coverage and payment for products under government health care programs. The ACA includes provisions of importance to our potential product candidate that:
• created an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic products, apportioned among these entities according to their market share in certain government healthcare programs;
• expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability;
• expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs and revising the definition of “average manufacturer price,” or AMP, for calculating and reporting Medicaid drug rebates on outpatient prescription drug prices;
88
• addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
• expanded the types of entities eligible for the 340B drug discount program;
• established the Medicare Part D coverage gap discount program by requiring manufacturers to provide point-of-sale-discounts off the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D; and
• created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the ACA. For example, the Tax Cuts and Jobs Act of 2017 (the Tax Act) was enacted, which includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court held in a 7–2 opinion that the states and individuals that brought the lawsuit challenging the ACA’s individual mandate do not have standing to challenge the law. The Supreme Court did not reach the merits of the challenge, but the decision ends the case. It is also unclear how the Supreme Court ruling, other such litigation, and the healthcare reform measures of the Biden administration will impact the ACA or our business.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2029 unless additional Congressional action is taken. The Coronavirus Aid, Relief, and Economic Security Act, or CARES Act, which was signed into law on March 27, 2020, designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended these reductions from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. In addition, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single-source and innovator multiple-source drugs, beginning January 1, 2024. These laws may result in additional reductions in Medicare, Medicaid and other healthcare funding.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, such as bundled payment models. In addition, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products, which has resulted in several Congressional inquiries and proposed and enacted state and federal legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing and importation. As a result, the FDA also released a final rule in September 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, in November 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions
89
reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed by the Biden administration until January 1, 2023. The CMS also issued an interim final rule that establishes a Most Favored Nation, or MFN, Model for Medicare Part B drug payments. This regulation would substantially change the reimbursement landscape as it bases Medicare Part B payment for 50 selected drugs on prices in foreign countries instead of average sales prices (ASP) and establishes a fixed add-on payment in place of the current 6 percent (4.3 percent after sequestration) of ASP. The MFN drug payment amount is expected to be lower than the current ASP-based limit because U.S. drug prices are generally the highest in the world. On December 28, 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. On January 13, 2021, in a separate lawsuit brought by industry groups in the U.S. District Court for the District of Maryland, the government defendants entered a joint motion to stay litigation on the condition that the government would not appeal the preliminary injunction granted in the U.S. District Court for the Northern District of California and that performance for any final regulation stemming from the MFN Model interim final rule shall not commence earlier than sixty (60) days after publication of that regulation in the Federal Register. In December 2020, CMS issued a final rule implementing significant manufacturer price reporting changes under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to pharmacy benefit manager accumulator programs and Best Price reporting related to certain value-based purchasing arrangements. On May 21, 2021, an industry group sued CMS, claiming that the change to the Best Price rule exceeds CMS’s statutory authority and is contrary to the Medicaid Rebate statute. This litigation is ongoing. It is unclear to what extent these new regulations will be implemented and to what extent these regulations or any future legislation or regulations by the Biden administration will have on our business, including our ability to generate revenue and achieve profitability.
On May 30, 2018, the Right to Try Act was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a drug manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act, but the manufacturer must develop an internal policy and respond to patient requests according to that policy.
Outside the United States, ensuring coverage and adequate payment for a product also involves challenges. Pricing of prescription pharmaceuticals is subject to government control in many countries. Pricing negotiations with government authorities can extend well beyond the receipt of regulatory approval for a product and may require a clinical trial that compares the cost-effectiveness of a product to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in commercialization.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies or so-called health technology assessments, in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a product or they may instead adopt a system of direct or indirect controls on our profitability for placing the product on the market. Other member states allow companies to fix their own prices for products, but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the European Union have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. The downward pressure on healthcare costs in general, particularly prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states, and parallel trade, i.e., arbitrage between low-priced and high-priced member states, can further reduce prices. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any products, if approved in those countries.
90
Compliance with other federal and state laws or requirements; changing legal requirements
If any products that we may develop are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, labeling, packaging, distribution, sales, promotion and other activities also are potentially subject to federal and state consumer protection and unfair competition laws, among other requirements to we may be subject.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with any of these laws or regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, exclusion from federal healthcare programs, requests for recall, seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, relabeling or repackaging, or refusal to allow a firm to enter into supply contracts, including government contracts. Any claim or action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Prohibitions or restrictions on marketing, sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling or packaging; (iii) the recall or discontinuation of our product candidates; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
Other U.S. environmental, health and safety laws and regulations
We may be subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and drug materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
We maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees, but this insurance may not provide adequate coverage against potential liabilities. However, we do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
Government regulation of drugs outside of the United States
To market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials, marketing authorization or identification of an alternate regulatory pathway, manufacturing, commercial sales and distribution of our product candidates. For instance, in the European Economic Area, or the EEA (comprised of the 26 EU Member States plus Iceland, Liechtenstein and Norway, with the UK having left the EU in January of 2020), medicinal products must be authorized for marketing by using either the centralized authorization procedure or national authorization procedures.
• Centralized procedure — If pursuing marketing authorization of a product candidate for a therapeutic indication under the centralized procedure, following the opining of the EMA’s Committee for Medicinal Products for Human Use, or, CHMP, the European Commission issues a single marketing authorization
91
valid across the EEA. The centralized procedure is compulsory for human medicines derived from biotechnology processes or advanced therapy medicinal products (such as gene therapy, somatic cell therapy and tissue engineered products), products that contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune diseases and other immune dysfunctions, viral diseases, and officially designated orphan medicines. For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the medicine concerned contains a new active substance not yet authorized in the EEA, is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health in the EEA. Under the centralized procedure the maximum timeframe for the evaluation of an MAA by the EMA is 210 days, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP. Accelerated assessment might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, particularly from the point of view of therapeutic innovation. The timeframe for the evaluation of an MAA under the accelerated assessment procedure is 150 days, excluding clock stops.
• National authorization procedures — There are also two other possible routes to authorize products for therapeutic indications in several countries, which are available for products that fall outside the scope of the centralized procedure:
• Decentralized procedure — Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EU country of medicinal products that have not yet been authorized in any EU country and that do not fall within the mandatory scope of the centralized procedure.
• Mutual recognition procedure — In the mutual recognition procedure, a medicine is first authorized in one EU Member State, in accordance with the national procedures of that country. Following this, additional marketing authorizations can be sought from other EU countries in a procedure whereby the countries concerned recognize the validity of the original, national marketing authorization.
In the EEA, new products for therapeutic indications that are authorized for marketing (i.e., reference products) qualify for eight years of data exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period prevents generic or biosimilar applicants from relying on the preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU during a period of eight years from the date on which the reference product was first authorized in the EU. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until ten years have elapsed from the initial authorization of the reference product in the EU. The ten-year market exclusivity period can be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
Similar to the United States, the various phases of non-clinical and clinical research in the European Union are subject to significant regulatory controls.
The Clinical Trials Directive 2001/20/EC, the Directive 2005/28/EC on GCP and the related national implementing provisions of the individual EU Member States govern the system for the approval of clinical trials in the European Union. Under this system, an applicant must obtain prior approval from the competent national authority of the EU Member States in which the clinical trial is to be conducted. Furthermore, the applicant may only start a clinical trial at a specific study site after the competent ethics committee has issued a favorable opinion. The clinical trial application must be accompanied by, among other documents, an investigational medicinal product dossier (the Common Technical Document) with supporting information prescribed by Directive 2001/20/EC, Directive 2005/28/EC, where relevant the implementing national provisions of the individual EU Member States and further detailed in applicable guidance documents.
In April 2014, the new Clinical Trials Regulation, (EU) No 536/2014 (Clinical Trials Regulation) was adopted. The Clinical Trials Regulation will be directly applicable in all the EU Member States, repealing the current Clinical Trials Directive 2001/20/EC. Conduct of all clinical trials performed in the European Union will continue to be bound by currently applicable provisions until the new Clinical Trials Regulation becomes applicable. The extent to which ongoing
92
clinical trials will be governed by the Clinical Trials Regulation will depend on when the Clinical Trials Regulation becomes applicable and on the duration of the individual clinical trial. If a clinical trial continues for more than three years from the day on which the Clinical Trials Regulation becomes applicable the Clinical Trials Regulation will at that time begin to apply to the clinical trial. The new Clinical Trials Regulation aims to simplify and streamline the approval of clinical trials in the European Union. The main characteristics of the regulation include: a streamlined application procedure via a single-entry point, the Clinical Trials Information System, or CTIS, a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. Part I is assessed by the competent authorities of all EU Member States in which an application for authorization of a clinical trial has been submitted (Member States concerned). Part II is assessed separately by each Member State concerned. Strict deadlines have been established for the assessment of clinical trial applications. The role of the relevant ethics committees in the assessment procedure will continue to be governed by the national law of the concerned EU Member State. However, overall related timelines will be defined by the Clinical Trials Regulation. On the basis of the results of an independent audit of the CTIS, on April 21, 2021, the EMA Management Board confirmed to the European Commission that the CTIS is fully functional. Based on this, January 31, 2022 was fixed as the date of applicability of the Clinical Trials Regulation (EU) No 536/2014.
The collection and use of personal health data in the European Union, previously governed by the provisions of the Data Protection Directive, is now governed by the General Data Protection Regulation, or the GDPR, which became effective on May 25, 2018. While the Data Protection Directive did not apply to organizations based outside the EU, the GDPR has expanded its reach to include any business, regardless of its location, that provides goods or services to residents in the EU. This expansion would incorporate any clinical trial activities in EU member states. The GDPR imposes strict requirements on controllers and processors of personal data, including special protections for “sensitive information” which includes health and genetic information of data subjects residing in the EU. GDPR grants individuals the opportunity to object to the processing of their personal information, allows them to request deletion of personal information in certain circumstances, and provides the individual with an express right to seek legal remedies in the event the individual believes his or her rights have been violated. Further, the GDPR imposes strict rules on the transfer of personal data out of the European Union to the United States or other regions that have not been deemed to offer “adequate” privacy protections. Failure to comply with the requirements of the GDPR and the related national data protection laws of the European Union Member States, which may deviate slightly from the GDPR, may result in fines of up to 4% of global revenues, or €20,000,000, whichever is greater. As a result of the implementation of the GDPR, we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules.
There is significant uncertainty related to the manner in which data protection authorities will seek to enforce compliance with GDPR. For example, it is not clear if the authorities will conduct random audits of companies doing business in the EU, or if the authorities will wait for complaints to be filed by individuals who claim their rights have been violated. Enforcement uncertainty and the costs associated with ensuring GDPR compliance are onerous and may adversely affect our business, financial condition, results of operations and prospects.
Should we utilize third party distributors, compliance with such foreign governmental regulations would generally be the responsibility of such distributors, who may be independent contractors over whom we have limited control.
Facilities
We lease a facility containing of approximately 4100 square feet of office space for our principal office, which is located at 61 Wilton Road on both the 3rd and 1st Floors, Westport CT, 06896. In September 2020 we signed a new 3 year lease for these private offices from a Landlord with an option to renew. We also sublease a portion of our space to a related party investor Portage Biotech. We believe that our facilities are adequate to meet our current needs and that suitable additional or substitute space at commercially reasonable terms will be available as needed to accommodate any future expansion of our operations.
Commercialization
We intend to pursue the complete development to our product candidates and, if marketing approval is obtained, to commercialize our product candidates on our own, or potentially with a partner, in the United States and other regions. We currently have no sales, marketing or commercial product distribution capabilities and have no experience as a company commercializing products. However, if necessary, we intend to hire appropriately to build the necessary
93
infrastructure and capabilities over time for the United States, and potentially other regions, following further advancement of our product candidates. Clinical data, the size of the addressable patient population, the size of the commercial infrastructure and manufacturing needs may all influence or alter our commercialization plans.
Manufacturing
We have established an operations leadership team with extensive experience in manufacturing drugs based on amphiphilic agents, and in the construction, validation, approval and operation of facilities designed to manufacture these products. We have established an operations leadership team with extensive experience in manufacturing of the SHAO and INT230-6 product candidate. Our team has developed a reproducible manufacturing process for SHAO and our product candidates. In 2016 we produced our first batch of INT230-6 under FDA regulated current Good Manufacturing Practice (cGMP) and have scaled up the product successfully. We generated and continue to generate stability data showing that INT230-6 had acceptable stability through 36 months using validated analytical methods.
Competition
The development and commercialization of new product candidates is highly competitive. We face competition from major pharmaceutical, specialty pharmaceutical and biotechnology companies among others with respect to INT230-6 and will face similar competition with respect to any product candidates that we may seek to develop or commercialize in the future. We compete in pharmaceutical, biotechnology and other related markets that develop immune-oncology therapies for the treatment of cancer. There are other companies working to develop new drugs, immunotherapies and other approaches for the treatment of cancer including divisions of large pharmaceutical and biotechnology companies of various sizes. The large pharmaceutical and biotechnology companies that have commercialized and/or are developing immune-based treatments for cancer include AstraZeneca, Bristol-Myers Squibb, Gilead Sciences, Inc., Merck & Co., Novartis, Pfizer and Genentech, Inc. In addition, other companies have oncology divisions including large companies such as Eli Lilly and GlaxoSmithKline or and several smaller midsize organizations.
Some of the products and therapies developed by our competitors are based on scientific approaches that are the similar to our approach, including with respect to the use of intratumoral delivery or activation of the immune system. Other competitive products and therapies are based on entirely different approaches. We are aware that Oncorus, Inc., Replimune Group, Inc., Amgen Inc., ImmVira Co., Ltd., IconOVir Bio, Inc., and FerGene, Inc., among others, are developing immunotherapies that may have utility for the treatment of indications that we are targeting. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Many of the companies we compete against or may compete against in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in concentration of even more resources among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, in establishing clinical trial sites and enrolling subjects for our clinical trials and in acquiring technologies complementary to, or necessary for, our programs.
We could see a reduction or elimination of our commercial opportunity if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, or are more convenient or are less expensive than any products that we or our collaborators may develop. Our competitors also may obtain FDA or foreign regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. The key competitive factors affecting the success of all our product candidates, if approved, are likely to be their efficacy, safety, convenience and price, if required, the level of biosimilar or generic competition and the availability of reimbursement from government and other third-party payors.
94
Intellectual Property
We are working to establish an intellectual property portfolio of both general know-how, issued patents and filed patent applications. We currently have three United States Patent and Trademark Office (PTO) issued patents; US Patent Number 9,351,997 is directed to a method of treating cancer, with a registration date of May 31, 2016 and an expiration date of December 6, 2033. US Patent Number 9,636,406 is directed to a method of treating cancer, with a registration date of May 2, 2017 and an expiration date of September 15, 2033. US Patent Number 10,888,618 is directed to a method of treating cancer, with a registration date of January 12, 2021 and an expiration date of September 15, 2033. One U.S. patent application is pending. US Patent Application Number 17/108,099 is directed to a method of treating cancer, with a filing date of December 1, 2020. We are prosecuting patents in every major market and have been granted patents in Australia, Canada, China, the 27 European Union countries (national phase filings were made for Austria, Belgium, Cypress, Czech Republic, Denmark, Finland, France, Germany, Greece, Italy, Ireland, Liechtenstein, Luxembourg, Macedonia, Malta, Monaco, the Netherlands, Norway, Poland, Portugal, Romania, San Marino, Spain, Sweden, Switzerland, Turkey, and the United Kingdom), Israel, Japan, Macau, Russia, South Africa, and South Korea. Patent applications are pending in Brazil, Chile, Mexico, India and Singapore.
Each application and issued patent has multiple claims directed to technology, methods, formulations and our lead product candidates. Together with trade secrets, know-how and continuing technological innovation, we believe that our IP position is thorough, novel, non-obvious and has been reduced to practice. The technology underlying the pending patent application directed to our lead product candidates has been developed by us and not acquired from in-licensing from any third party.
Employees and Human Capital Resources
As of January 7, 2022, we had 15 employees and contractors, including four with M.D. and/or Ph.D. degrees. There were six full time employees and two part time employee. We have never had a work stoppage, and none of our employees is represented by a labor organization or under any collective-bargaining arrangements. We consider our employee relations to be good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and our success by motivating such individuals to perform to the best of their abilities and achieve our objectives.
Legal Proceedings
From time to time, we may become involved in litigation or other legal proceedings. We are not currently nor have we ever been a party to any litigation or legal proceedings that, in the opinion of our management, are probable to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on our business, financial condition, results of operations and prospects because of defense and settlement costs, diversion of management resources and other factors.
95
The following table sets forth the name, age (as of January 7, 2022) and position of individuals who currently serve as our directors and executive officers. The following also includes certain information regarding our directors’ and officers’ individual experience, qualifications, attributes and skills, and brief statements of those aspects of our directors’ backgrounds that led us to conclude that they should serve as directors.
|
Name |
Age |
Position |
||
|
Lewis H. Bender |
62 |
President, Chief Executive Officer and Chairman of the Board |
||
|
Dr. Ian B. Walters |
54 |
Vice President and Chief Medical Officer |
||
|
James M. Ahlers |
57 |
Chief Financial Officer |
||
|
John Wesolowski |
62 |
Principal Accounting Officer and Controller |
||
|
Rebecca Drain |
58 |
Vice President, Regulatory Affairs and Quality |
||
|
Dr. Declan Doogan |
69 |
Director |
||
|
Dr. Emer Leahy |
55 |
Director |
||
|
Dr. Mark A. Goldberg |
61 |
Director |
Executive Officers
Lewis H. Bender is our founder and has served as our President and Chief Executive Officer since April 2012. Prior to our founding, Mr. Bender was the CEO of publicly traded (AMEX & OTC) Interleukin Genetics, Inc. from 2008 until 2012. Interleukin was a personalized medicine company. Mr. Bender was successful in raising capital for us via a direct placement with institutional investors and partnered with the insurance industry for development of an IG product. Prior to joining Interleukin Genetics, Mr. Bender held numerous positions at Emisphere Technologies, Inc. at the time a publicly traded (Nasdaq) drug delivery company specializing in the development of oral delivery of poorly absorbed molecules. While at Emisphere from 1993 to December 2007, Mr. Bender held positions including Interim President & CEO, Chief Technology Officer, Senior Vice President of Business Development, and Vice President of Manufacturing and Process Development. Mr. Bender has over 26 years of biotech and pharmaceutical executive management experience. He has led development teams taking products from discovery to Phase 3 for compounds using novel drug delivery techniques. Mr. Bender has a both a BS and MS in Chemical Engineering from The Massachusetts Institute of Technology (MIT), an MBA from the University of Pennsylvania’s Wharton School, and an MA in International Studies also from the University of Pennsylvania. He is fluent in French and German.
Dr. Ian B. Walters has served as our Vice President and Chief Medical Officer since August 2014. Dr. Walters brings over 15 years of oncology/immunology drug development experience to our team. Dr. Walters has clinical development experience with 30+ compounds, and has been a consultant to biotech, pharma and investment companies specializing in the evaluation, prioritization, and development of innovative technologies in the treatment of severe diseases. He has worked in multiple biotech companies on corporate development, translational medicine, clinical development and medical affairs including BMS, Millennium, PDL and the Rockefeller University. Dr. Walters spent seven years at Bristol-Myers Squibb (BMS), where he led clinical research and matrix development teams. During his tenure there he contributed to the development of multiple immuno-oncology products, Yervoy® (ipilimumab) and Opdivo (nivolumab, an anti-PD-1 drug), as well as the licensing and partnering strategy for other immuno-oncologic agents. Before entering the private sector, Dr. Walters was a lead investigator at the Rockefeller University and initiated cutting edge immunology research to understand the mechanism of action of several compounds. Dr. Walters received his MD from the Albert Einstein College of Medicine and an MBA from the Wharton School of The University of Pennsylvania.
James M. Ahlers has served as Chief Financial Officer since January 2022 through a consulting agreement with Mr. Ahlers’ employer, Danforth Advisors LLC, a company that provides strategic and operational finance and accounting services to life science companies. From February 2002 to November 2019 Mr. Ahlers served as Chief Financial Officer of Intarcia Therapeutics, Inc. Mr. Ahlers is an accomplished finance leader with 25 years of experience building life science businesses. During his career, he has managed capital raising transactions, including IPOs, that have raised in excess of $2 billion. In addition, he has developed and implemented international operations and global tax strategies. Mr. Ahlers holds a B.S. in accounting from the University of San Francisco. Mr. Ahlers replaces Greg Wade, who left Danforth at the end of 2021 to pursue a full time position in business development at a third party biotech firm.
96
John Wesolowski has served as our Principal Accounting Officer and Controller since March 2017. Prior to joining Intensity Therapeutics, from 1998 to 2016 Mr. Wesolowski was Director of Costing in the Yale University Controller’s office. In that role Mr. Wesolowski conducted financial reporting, property tax management, was responsible for calculations of overhead and benefit rates, and was involved in numerous special projects related to accounting process and controls. Also, at Yale, he was involved in financial reporting and the accounting matters related to clinical trials and other organized research. Prior to joining Yale Mr. Wesolowski was the Vice President and Controller for Automatic Fastener Corporation in Branford, CT from 1988 to 1998. In this role, Mr. Wesolowski oversaw all accounting, purchasing and human resource functions. John also has 5 years of experience in public accounting and auditing from working at KMG Main Hurdman, now KPMG. Mr. Wesolowski received a Bachelor of Science in Finance from The Pennsylvania State University (Penn State at University Park) and an MBA from the University of Connecticut in Management Science. He is a Certified Public Account since 1983.
Rebecca Drain has served as our Vice President, Regulatory Affairs and Quality since August 2021 and prior to that as our Executive Director of Regulatory Affairs and Quality Assurance since July 2019. Prior to joining Intensity Therapeutics, Ms. Drain has over 25 years of experience with Bristol Myers Squibb, where most recently, between January 2015 and December 2018, she served as Director, Submission Management, Global Regulatory Safety and Biometrics, a position in which she was responsible for global oncology regulatory submissions. Prior to BMS, she was a research scientist with a drug discovery company. Ms. Drain earned a B.S. in Pathobiology from the University of Connecticut.
Non-Employee Directors
Dr. Declan Doogan, M.D. has served as a member of our board of directors since June 2016. Dr. Doogan is a seasoned drug development executive and life sciences investor with more than 30 years’ experience in the global pharmaceutical industry in both major pharmaceutical and biotechnology companies. Since November 2019 he has served as Chief Medical Officer at Juvenescence Ltd. Since February 2021, he has served as a director of Tenax Therapeutics, Inc. (NASDAQ:TENX). Since June 2013, Dr. Doogan has served as a director of Portage Biotech, Inc. (NASDAQ:PRTG), where he also served as chief executive officer from June 2013 through May 2019. He is a co-founder of Biohaven Pharmaceuticals (NYSE:BHVN), has served as a member of its board of directors since its inception in September 2013 and currently serves as its chairman. He was a director of Sosei Group Corporation from June 2007 to June 2019. Starting in 2007, Dr. Doogan also held executive roles at Amarin (AMRN: Nasdaq), where he initially served as Head of Research and Development and then, from 2009 until 2010, as Interim Chief Executive Officer and then as Chief Medical Officer until 2012. He joined Pfizer in 1982, where he held a number of senior positions in R&D in the USA, UK and Japan, laterally as the Senior Vice President and Head of Worldwide Development until leaving in 2007. Beyond his executive career, Dr. Doogan is an investor in emerging biotechnology companies, and is a partner at Mediqventures. Dr. Doogan has also held professorships at Harvard School of Public Health, Glasgow University Medical School and Kitasato University (Tokyo). He received his medical degree from Glasgow University in 1975. He is a Fellow of the Royal College of Physicians and the Faculty Pharmaceutical Medicine and holds a Doctorate of Science at the University of Kent in the UK. We believe that Dr. Doogan’s extensive operational experience in the pharmaceutical and biotech industries qualifies him to serve as a member of our Board.
Dr. Emer Leahy has served on our board of directors since June 2016. Dr. Leahy received her Ph.D. in Neuropharmacology from University College Dublin, Ireland, and her MBA from Columbia University. Since 2000, she has served as CEO of PsychoGenics Inc., a profitable preclinical CNS service company. She is also CEO of PGI Drug Discovery LLC, a company engaged in psychiatric drug discovery with five partnered clinical programs including one in Phase 3. Further, she holds an Adjunct Associate Professor of Neuroscience position at Mount Sinai School of Medicine. Dr. Leahy has more than 30 years of experience in drug discovery, clinical development and business development for pharmaceutical and biotechnology companies, including extensive knowledge of technology assessment, licensing, mergers and acquisitions, and strategic planning. Dr. Leahy served on the Emerging Companies Section Governing Board for the Board of Directors of the Biotechnology Industry Organization (BIO), the Business Review Board for the Alzheimer’s Drug Discovery Foundation, and the Scientific Advisory Board of the International Rett Syndrome Foundation. She also currently serves on the Board of Directors of PsychoGenics Inc., Bright Minds Biosciences, Pasithea Therapeutics, and on the Board of Trustees of BIONJ. We believe that Dr. Leahy’s extensive experience in the biopharmaceutical industry, including as a CEO of several companies, allows her to make valuable contributions to the Board.
97
Dr. Mark A. Goldberg has served as a member of our board of directors since May 2018. Dr. Mark A. Goldberg recently served as President and COO of PAREXEL International, one of the world’s largest global biopharmaceutical service providers, with consolidated service revenue of approximately $2.1 billion, over 18,000 employees, and 86 locations in 51 countries. He was responsible for overseeing all revenue generating business segments including Clinical Research Services, PAREXEL Informatics, and PAREXEL Consulting as well as sales, marketing, corporate quality, and information technology. Dr. Goldberg helped to pioneer PAREXEL’s strategic partnering approach with some of the world’s leading pharmaceutical companies and to build out the company’s global infrastructure, particularly in the Asia Pacific region, through both organic growth and acquisitions. Earlier in his PAREXEL career, he founded the company’s Medical Imaging business and helped establish its technology subsidiary, Perceptive Informatics (now PAREXEL Informatics). Dr. Goldberg holds a BS degree in computer science from MIT and an MD from the University of Massachusetts Medical School. He completed residency training in Radiology at Massachusetts General Hospital, where he also served as Chief Resident and a staff physician with academic appointments at Harvard Medical School. We believes that Dr. Goldberg’s medical background and public company board experience allows him to make valuable contributions to our Board.
Board Composition and Election of Directors
Our business and affairs are managed under the direction of our board of directors. The primary responsibilities of our board of directors are to provide oversight, strategic guidance, counseling and direction to our management. Our board of directors meets on a regular basis and additionally as required.
The number of directors will be fixed by our board of directors, subject to the terms of our amended and restated certificate of incorporation and our amended and restated bylaws that will become effective immediately prior to the completion of this offering. Our board of directors consists of four (4) directors, three (3) of whom will qualify as “independent” under Nasdaq listing standards.
Directors will (except for the filling of vacancies and newly created directorships) be elected by the holders of a plurality of the votes cast by the holders of shares present in person or represented by proxy at the meeting and entitled to vote on the election of such directors. In accordance with our amended and restated certificate of incorporation and our amended and restated bylaws, which will become effective immediately prior to the completion of this offering, immediately after the completion of this offering our board of directors will be divided into three classes with staggered three-year terms. Only one class of directors will be elected at each annual meeting of our stockholders, with the other classes continuing for the remainder of their respective three-year terms. Our directors will be divided among the three classes as follows:
• the Class I director will be Dr. Declan Doogan, and his term will expire at the first annual meeting of stockholders after the completion of this offering;
• the Class II director will be Dr. Mark A. Goldberg, and his term will expire at the second annual meeting of stockholders after the completion of this offering; and
• the Class III directors will be Dr. Emer Leahy and Lewis H. Bender, and their terms will expire at the third annual meeting of stockholders after the completion of this offering.
Each director’s term will continue until the election and qualification of his or her successor, or his or her earlier death, resignation or removal. Any increase or decrease in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors. This classification of our board of directors may have the effect of delaying or preventing changes in control of our company.
Director Independence
Our board of directors has undertaken a review of the independence of each director. Based on information provided by each director concerning his or her background, employment and affiliations, our board of directors has determined that Dr. Declan Doogan, Dr. Emer Leahy and Dr. Mark A. Goldberg do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the applicable rules and regulations of the SEC and the listing standards of Nasdaq. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our board
98
of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director, and the transactions involving them described in the section titled “Certain Relationships and Related Party Transactions.”
Committees of the Board of Directors
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. The composition and responsibilities of each of the committees of our board of directors is described below. Members will serve on these committees until their resignation or until as otherwise determined by our board of directors.
Audit Committee
Our audit committee consists of Dr. Declan Doogan, Dr. Emer Leahy and Dr. Mark A. Goldberg, with Dr. Emer Leahy serving as Chairperson. The composition of our audit committee meets the requirements for independence under current Nasdaq listing standards and SEC rules and regulations. Each member of our audit committee meets the financial literacy requirements of Nasdaq listing standards. In addition, our board of directors has determined that Dr. Emer Leahy is an audit committee financial expert within the meaning of Item 407(d) of Regulation S-K under the Securities Act of 1933. Our audit committee will, among other things:
• review our consolidated financial statements and our critical accounting policies and practices;
• select a qualified firm to serve as the independent registered public accounting firm to audit our consolidated financial statements;
• help to ensure the independence and performance of the independent registered public accounting firm;
• discuss the scope and results of the audit with the independent registered public accounting firm and review, with management and the independent registered public accounting firm, our interim and year-end results of operations;
• pre-approve all audit and all permissible non-audit services to be performed by the independent registered public accounting firm;
• oversee the performance of our internal audit function when established;
• review the adequacy of our internal controls;
• develop procedures for employees to submit concerns anonymously about questionable accounting or audit matters;
• review our policies on risk assessment and risk management; and
• review related party transactions.
Our audit committee operates under a written charter, to be effective prior to the completion of this offering, that satisfies the applicable rules of the SEC and the listing standards of Nasdaq.
Compensation Committee
Our compensation committee consists of Dr. Declan Doogan, Dr. Emer Leahy and Dr. Mark A. Goldberg, with Dr. Declan Doogan serving as Chairperson. The composition of our compensation committee meets the requirements for independence under Nasdaq listing standards and SEC rules and regulations. Each member of the compensation committee is also a non-employee director, as defined pursuant to Rule 16b-3 promulgated under the Exchange Act. The purpose of our compensation committee is to discharge the responsibilities of our board of directors relating to compensation of our executive officers. Our compensation committee will, among other things:
• review, approve and determine, or make recommendations to our board of directors regarding, the compensation of our executive officers;
• administer our stock and equity incentive plans;
99
• review and approve, or make recommendations to our board of directors regarding, incentive compensation and equity plans; and
• establish and review general policies relating to compensation and benefits of our employees.
Our compensation committee will operate under a written charter, to be effective prior to the completion of this offering, that satisfies the applicable rules of the SEC and the listing standards of Nasdaq.
Nominating and Corporate Governance Committee
Immediately following the completion of this offering, our nominating and corporate governance committee will consist of Dr. Declan Doogan and Dr. Mark A. Goldberg, with Dr. Mark A. Goldberg serving as Chairperson. The composition of our corporate governance committee meets the requirements for independence under Nasdaq listing standards and SEC rules and regulations. Our nominating and corporate governance committee will, among other things:
• identify, evaluate and select, or make recommendations to our board of directors regarding, nominees for election to our board of directors and its committees;
• evaluate the performance of our board of directors and of individual directors;
• consider and make recommendations to our board of directors regarding the composition of our board of directors and its committees;
• review developments in corporate governance practices;
• oversee environmental, social and governance (ESG) matters;
• evaluate the adequacy of our corporate governance practices and reporting; and
• develop and make recommendations to our board of directors regarding corporate governance guidelines and matters.
The nominating and corporate governance committee will operate under a written charter, to be effective prior to the completion of this offering, that satisfies the applicable listing requirements and rules of Nasdaq.
Role of Board of Directors in Risk Oversight Process
Our board of directors has responsibility for the oversight of our risk management processes and, either as a whole or through its committees, regularly discusses with management our major risk exposures, their potential impact on our business and the steps we take to manage them. The risk oversight process includes receiving regular reports from board committees and members of senior management to enable our board of directors to understand our risk identification, risk management and risk mitigation strategies with respect to areas of potential material risk, including operations, finance, legal, regulatory, cybersecurity, strategic and reputational risk.
Code of Business Conduct
Upon completion of this offering, our board of directors will establish a Code of Conduct applicable to our directors, officers and employees. The Code of Conduct will be accessible on our website at www.intensitytherapeutics.com. If we make any substantive amendments to the Code of Conduct or grant any waiver, including any implicit waiver, from a provision of the Code of Conduct to our officers, we will disclose the nature of such amendment or waiver on that website or in a report on Form 8-K.
Compensation Committee Interlocks and Insider Participation
All compensation and related matters are reviewed by our compensation committee. Our compensation committee consists of consists of Dr. Declan Doogan, Dr. Emer Leahy and Dr. Mark A. Goldberg, with Dr. Declan Doogan serving as Chairperson. None of the members of our compensation committee is or has at any time during the past year been an officer or employee of ours. None of our executive officers currently serves or in the past year has served as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving on our board of directors or
100
Our named executive officers, or NEOs, for the year ended December 31, 2021, which consist of each person who served as our principal executive officer during 2021 and the next three most highly compensated executive officers, are:
• Lewis H. Bender, President and Chief Executive Officer
• Rebecca Drain, Vice President, Regulatory Affairs and Quality
• Dr. Ian B. Walters, Vice President and Chief Medical Officer
Executive Compensation Overview
To date, the compensation of our NEOs has primarily consisted of a combination of base salary and long-term incentive compensation in the form of stock options. Our NEOs, like all full-time employees, are eligible to participate in our health and dental benefit plans and 401(k) plan matching program. As we transition from a private company to a publicly traded company, we intend to evaluate our compensation values and philosophy and compensation plans and arrangements as circumstances require. At a minimum, we expect to review executive compensation annually with input from a compensation consultant. As part of this review process, we expect the board of directors and the compensation committee to apply our values and philosophy, while considering the compensation levels needed to ensure our executive compensation program remains competitive. We will also review whether we are meeting our retention objectives and the potential cost of replacing a key employee.
2021 Summary Compensation Table
The following table presents all of the compensation awarded to or earned by our named executive officers for the year ended December 31, 2021.
|
Name and Principal Position |
YEAR |
SALARY |
BONUS |
STOCK |
OPTION |
NON-EQUITY |
ALL OTHER |
TOTAL |
||||||||
|
Lewis H. Bender |
2021 |
434,255 |
— |
— |
581,349 |
— |
62,412 |
1,078,016 |
||||||||
|
President and Chief Executive Officer |
||||||||||||||||
|
Rebecca Drain |
2021 |
226,923 |
— |
— |
96,892 |
— |
55,106 |
378,921 |
||||||||
|
Vice President, Regulatory Affairs and Quality |
||||||||||||||||
|
Dr. Ian B. Walters |
2021 |
113,397 |
40,073 |
— |
193,783 |
— |
50,179 |
397,432 |
||||||||
|
Vice President and Chief Medical Officer |
____________
(1) In accordance with SEC rules, these columns reflect the aggregate grant date fair value of the option awards and stock awards granted during 2021 computed in accordance with Financial Accounting Standard Board ASC Topic 718 for stock-based compensation transactions, or ASC 718. Assumptions used in the calculation of these amounts are exercise price of $5.75, expected term of 4 years, risk free interest rate of 1.04%, and expected volatility of 96.84%. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of stock options, the exercise of stock options or the sale of shares of our common stock. During 2021, Mr. Bender, Ms. Drain and Dr. Walters were granted options to purchase 150,000, 25,000 and 50,000 shares of common stock, respectively.
(2) The following table provides information regarding the compensation disclosed in the All Other Compensation column. This information includes identification and quantification of each perquisite and personal benefit received by each NEO, regardless of amount.
|
Medical |
401K |
Perquisites and |
Total |
|||||
|
Lewis H. Bender |
51,099 |
8,700 |
2,613 |
62,412 |
||||
|
Rebecca Drain |
48,298 |
6,808 |
— |
55,106 |
||||
|
Dr. Ian B. Walters |
46,777 |
3,402 |
— |
50,179 |
____________
(1) Represents company-paid portion of health and dental insurance.
(2) Represents matching 401(k) Plan contributions of up to 3% of eligible earnings.
(3) Executive perquisites and personal benefits include cell phone and home internet service.
101
Narrative Disclosure to the Summary Compensation Table
Annual Base Salary
Our NEOs each receive a base salary to compensate them for services rendered to our company. The base salary payable to each named executive officer is intended to provide a fixed component of compensation reflecting the executive’s skill set, experience, role and responsibilities. Base salaries are reviewed annually, typically in connection with our annual performance review process, approved by our board of directors or the compensation committee, and may be adjusted from time to time to realign salaries with market levels after taking into account individual responsibilities, performance, and experience.
For fiscal year 2021, the annual base salary for each of Mr. Bender, Ms. Drain and Dr. Walters were $434,255, $226,923, and $113,397, respectively.
All Other Compensation
All other compensation includes: 1) Stock options reflecting the fair value of stock options granted during 2021 in accordance with ASC Topic 718; 2) 401(k) plan matching contribution reflecting 3% of eligible earnings; and 3) allowance for cell phone and home office internet.
Outstanding Equity Awards at Fiscal Year End
The following table presents the outstanding equity awards held by each of our named executive officers as of December 31, 2021:
|
OPTION AWARDS(1) |
||||||||||||
|
Name |
VESTING |
NUMBER OF |
NUMBER OF |
EQUITY |
OPTION |
OPTION |
||||||
|
Lewis H. Bender |
8/6/2019 |
150,000 |
— |
4.50 |
8/6/2029 |
|||||||
|
7/31/2020 |
37,500 |
112,500 |
5.75 |
7/31/2030 |
||||||||
|
8/13/2021 |
37,500 |
112,500 |
5.75 |
8/13/2031 |
||||||||
|
Rebecca Drain |
7/11/2019 |
15,000 |
15,000 |
4.50 |
7/11/2029 |
|||||||
|
7/31/2020 |
3,125 |
9,375 |
5.75 |
7/31/2030 |
||||||||
|
8/13/2021 |
— |
25,000 |
5.75 |
8/13/2031 |
||||||||
|
Dr. Ian B. Walters |
8/25/2014 |
175,000 |
— |
1.50 |
8/25/2024 |
|||||||
|
9/21/2015 |
60,000 |
— |
2.00 |
9/21/2025 |
||||||||
|
6/24/2016 |
100,000 |
— |
2.00 |
6/24/2026 |
||||||||
|
9/9/2016 |
60,000 |
— |
2.00 |
9/9/2026 |
||||||||
|
2/6/2018 |
52,500 |
17,500 |
4.00 |
2/6/2028 |
||||||||
|
7/11/2019 |
7,500 |
7,500 |
4.50 |
7/11/2029 |
||||||||
|
7/31/2020 |
11,250 |
33,750 |
5.75 |
7/31/2030 |
||||||||
|
8/13/2021 |
— |
50,000 |
5.75 |
8/13/2031 |
||||||||
Employment Agreements
We have entered into an Amended and Restated Employment Agreement with Mr. Bender in connection with this offering (the “Amended and Restated Employment Agreement”), which agreement became effective on November 29, 2021.
The Amended and Restated Employment Agreement provides that Mr. Bender will receive a base salary of $523,000, which will be reviewed annually and may be increased, but not decreased, without the Mr. Bender’s consent. The Amended and Restated Employment Agreement also provides that Mr. Bender is eligible to receive an annual performance-based cash bonus as a percentage (not more than 75%) of base salary, which bonus is earned based on the achievement of performance targets, as determined annually by the Compensation Committee of our board of directors. Any annual bonus, to the extent earned, is paid in a lump sum. Under the Amended and Restated Employment
102
Agreement, Mr. Bender is also eligible to participate in the Company’s equity grant program, which grants shall occur not less than once per year. The form of equity award agreement and the terms and conditions of such equity awards, including with respect to vesting, will be determined by our board of directors.
Under the Amended and Restated Employment Agreement, Mr. Bender may terminate his employment at any time and for any reason with prior notice. We may terminate Mr. Bender’s employment immediately upon his death, upon a period of disability or immediately upon written notice for “cause” (as defined below). In the event that Mr. Bender’s employment is terminated due to his death or disability, for “cause” or upon his resignation without “good reason” (as defined below), we must provide him (or his beneficiaries) with (i) any unpaid base salary through the date of termination, (ii) payment for any accrued but unused paid time off, (iii) reimbursement for expenses properly incurred, and (iv) all other vested entitlements or benefits to which he is entitled (collectively, the “Accrued Benefits”).
If we terminate the executive’s employment without cause or Mr. Bender terminates his employment for “good reason” (as defined below), then we must provide Mr. Bender with the Accrued Benefits and subject to his execution and non-revocation of a release of claims, a lump sum payment equal to two times the sum of (i) his annual base salary, plus (ii) his target annual bonus, in each case at the rates and target amounts in effect as of such termination of employment. If we terminate the executive’s employment without cause or Mr. Bender terminates his employment for good reason and such termination is concurrent with or within six months after a change of control of the Company, then in addition to receiving the Accrued Benefits, but in lieu of other severance payments, Mr. Bender shall receive as a lump sum severance payment, at the time of such termination, an amount equal to (i) two and one-half (2.5) times the sum of (A) his base salary and (B) target annual bonus, each as in effect at the time of such termination, plus (ii) a payment equal to his target annual bonus for the calendar year in which the termination date occurred pro-rated for the period for which Mr. Bender was employed by us during such year.
For purposes of the Amended and Restated Employment Agreement, “cause” generally means the executive’s (i) the failure by the executive to cure a breach of a material duty imposed on the executive under the Amended and Restated Employment Agreement or any other written agreement between executive and the Company, or any policy of the Company, after written notice thereof by the Company, if curable in the reasonable discretion of the Board, (ii) acts by executive of fraud, embezzlement, theft, willful misconduct, gross negligence, or other material dishonesty directed against the Company, (iii) the failure or refusal by executive to perform any material duties under the Amended and Restated Employment Agreement or to follow any lawful and reasonable direction of the Company; or (vi) the executive’s being charged with a felony (other than a traffic offense), or a crime involving moral turpitude.
For purposes of the Amended and Restated Employment Agreement, “good reason” generally means a resignation by the executive on account of: (i) a material reduction in the executive’s duties, authority or responsibilities; (ii) relocation of executive’s place of employment without executive’s consent to a location more than fifty miles from the Company’s current executive offices; or (iii) any material breach by the Company of the Amended and Restated Employment Agreement. Good reason will not exist unless the executive notifies the Company in writing of such action not later than a set time after its initial occurrence and the Company has not remediated the action within a set time after such notice.
We previously entered into employment agreements with each of Rebecca Drain, effective June 21, 2019, and Dr. Ian B. Walters, effective August 25, 2014 (collectively, the “Employment Agreements”). Each of the Employment Agreements provides for at-will employment.
Each Employment Agreement provides the terms of compensation provided to each executive for their services. In the case of Ms. Drain, the Employment Agreement provides for a base annual salary of $200,000 and 30,000 incentive options from the Company’s equity incentive plan. In the case of Dr. Walters, the Employment Agreement provides for a monthly salary of $1,000 until the Company concludes a new financing of $1,500,000 or greater and thereafter the salary shall increase to $5,000. Dr. Walters’ Employment Agreement also includes a grant of up to 175,000 incentive options from the Company’s equity incentive plan.
Either we or the executive may terminate the respective Employment Agreement at any time for any or no reason, provided, however, that, at our request, the executive has agreed to continue as an employee for an additional thirty (30) day period after the termination date for the purpose of assisting with locating and training a suitable replacement.
The Employment Agreements also include customary confidentiality and non-disparagement provisions, as well as provisions relating to assignment of inventions. The Employment Agreements also includes non-competition and non-solicitation of employees and customers provision that run during the executive’s employment with the Company and for a period of one year after termination of employment.
103
Director Compensation
The following table provides certain information concerning compensation for each person who served as a non-employee member of our board of directors during the year ended December 31, 2021. Other than as set forth in the table and described more fully below, we did not pay any compensation, make any equity awards or non-equity awards to, or pay any other compensation to any of the non-employee members of our board of directors in 2021. During fiscal year 2021, Lewis H. Bender, our President and Chief Executive Officer, served as a member of our board of directors and received no additional compensation for his services as a member of our board of directors. See the section titled “Executive Compensation” for more information about Mr. Bender’s compensation for fiscal year 2021. We reimburse non-employee members of our board of directors for reasonable travel and out-of-pocket expenses incurred in attending meetings of our board of directors and committees of our board of directors.
|
Name |
FEES EARNED OR PAID IN CASH |
STOCK |
OPTION |
ALL OTHER |
TOTAL |
|||||
|
Dr. Declan Doogan |
— |
— |
193,783 |
— |
193,783 |
|||||
|
Dr. Emer Leahy |
— |
— |
193,783 |
— |
193,783 |
|||||
|
Dr. Mark A. Goldberg |
— |
— |
193,783 |
— |
193,783 |
Non-Employee Director Compensation Policy
Our board of directors has adopted a non-employee director compensation policy that is designed to enable us to attract and retain, on a long-term basis, highly qualified non-employee directors. Under the policy, each director who is not an employee will be paid cash compensation from and after the completion of this offering, as set forth below:
|
ANNUAL RETAINER |
|||
|
Board of Directors: |
|
||
|
All non-employee members |
$ |
40,000 |
|
|
Audit Committee: |
|
||
|
Chair |
$ |
20,000 |
|
|
Members |
$ |
10,000 |
|
|
Compensation Committee: |
|
||
|
Chair |
$ |
15,000 |
|
|
Members |
$ |
7,000 |
|
|
Corporate Governance Committee: |
|
||
|
Chair |
$ |
10,000 |
|
|
Members |
$ |
5,000 |
|
Compensation Plans
2013 Stock and Option Plan
Under our 2013 Stock and Option Plan, or the 2013 Plan, 4,500,000 shares of common stock have been reserved for issuance in the form of incentive stock options, non-qualified stock options, restricted stock, unrestricted stock, stock appreciation rights or any combination of the foregoing. The shares issuable pursuant to awards granted under the 2013 Plan are authorized but unissued shares.
The 2013 Plan is administered by our board or at the discretion of the board, which has full power to select the individuals to whom awards will be granted and to determine the specific terms and conditions of each award, subject to the provisions of the 2013 Plan. Pursuant to the 2013 Plan and subject to applicable law, our board of directors has delegated to the compensation committee the power to make recommendations to the board of directors relating to management compensation, the adoption of employee benefit plans, stock option or equity incentive plans and other similar matters.
The option exercise price of each option granted under the 2013 Plan is determined by our board of directors and may not be less than the fair market value of a share of common stock on the date of grant. The term of each option is fixed by the board and may not exceed 10 years from the date of grant. The board determines at what time or times each option may be exercised when granting the option.
104
The 2013 Plan provides that, upon the consummation of a sale event, unless provision is made in connection with the sale event for the assumption or continuation of the awards by the successor entity or substitution of the awards with new awards of the successor entity, with appropriate adjustment, the 2013 Plan and all outstanding and unexercised options issued thereunder will terminate upon the effective time of the sale event. We may make or provide for cash payment to holders of options equal to the difference between (i) the per share cash consideration in the sale event multiplied by the number of shares subject to outstanding options being cancelled, and (ii) the aggregate exercise price to the holders of all vested and exercisable options.
Our board of directors may amend the 2013 Plan but no such action may adversely affect the rights of an award holder without such holder’s consent. Approval by our stockholders of amendments to the 2013 Plan must be obtained if required by law.
As of December 31, 2021, options to purchase 1,822,500 shares of common stock were outstanding under the 2013 Plan. Our board of directors has determined not to make any further awards under the 2013 Plan following the closing of this offering.
2021 Stock Incentive Plan
In connection with this offering, we plan to adopt a new equity incentive plan, the 2021 Stock Incentive Plan, or the 2021 Plan. Under the 2021 Plan, we may grant cash and equity incentive awards to eligible service providers in order to attract, motivate and retain the talent for which we compete. The material terms of the 2021 Plan are summarized below.
Types of Awards. The 2021 Plan provides for the grant of non-qualified stock options (“NQSOs”), incentive stock options (“ISOs”), restricted stock awards, restricted stock units (“RSUs”), unrestricted stock awards, stock appreciation rights and other forms of stock based compensation.
Eligibility and Administration. Employees, officers, consultants directors, and other service providers of the Company and its affiliates are eligible to receive awards under the 2021 Plan. The 2021 Plan is administered by the board with respect to awards to non-employee directors and by the Compensation Committee with respect to other participants, each of which may delegate its duties and responsibilities to committees of the company’s directors and/or officers (all such bodies and delegates referred to collectively as the plan administrator), subject to certain limitations that may be imposed under Section 16 of the Exchange Act, and/or other applicable law or stock exchange rules, as applicable. The plan administrator has the authority to make all determinations and interpretations under, prescribe all forms for use with, and adopt rules for the administration of, the 2021 Plan, subject to its express terms and conditions. The plan administrator also sets the terms and conditions of all awards under the 2021 Plan, including any vesting and vesting acceleration conditions.
Share Reserve. Pursuant to the 2021 Plan, we have reserved 3,000,000 shares of the Common Stock for issuance thereunder, which reserve shall be increased annually beginning on January 1, 2022 and ending on and including January 1, 2031, equal to the lesser of (A) 3.5% of the aggregate number of shares of Common Stock outstanding on the final day of the immediately preceding calendar year or (B) such smaller number of shares as is determined by our board. The share reserve is subject to the following adjustments:
• The share limit is increased by the number of shares subject to awards granted that later are forfeited, expire or otherwise terminate without issuance of shares, or that are settled for cash or otherwise do not result in the issuance of shares.
• Shares that are withheld upon exercise to pay the exercise price of a stock option or satisfy any tax withholding requirements are added back to the share reserve and again are available for issuance under the 2021 Plan.
Awards issued in substitution for awards previously granted by a company that merges with, or is acquired by, the Company do not reduce the share reserve limit under the 2021 Plan.
Director Compensation. The 2021 Plan provides for an annual limit on non-employee director compensation of $500,000, increased to $750,000 in the fiscal year of a non-employee director’s initial service as a non-employee member of the board of directors of the Company. This limit applies to the sum of both equity grants that could be awarded to non-employee directors during a fiscal year (based on their value under ASC Topic 718 on the grant date)
105
and cash compensation, such as cash retainers and meeting fees earned during a fiscal year. Notwithstanding the foregoing, the board reserves the right to make an exception to these limits due to extraordinary circumstances without the participation of the affected director receiving the additional compensation.
Stock Options. ISOs may be granted only to employees of the Company, or to employees of a parent or subsidiary of the Company, determined as of the date of grant of such options. An ISO granted to a prospective employee upon the condition that such person becomes an employee shall be deemed granted effective on the date such person commences employment. The exercise price of an ISO shall not be less than 100% of the fair market value of the shares covered by the awards on the date of grant of such option or such other price as may be determined pursuant to the Internal Revenue Code of 1986, as amended from time to time (the “Code”). Notwithstanding the foregoing, an ISO may be granted with an exercise price lower than the minimum exercise price set forth above if such award is granted pursuant to an assumption or substitution for another option in a manner that complies with the provisions of Section 424(a) of the Code. Notwithstanding any other provision of the 2021 Plan to the contrary, no ISO may be granted under the 2021 Plan after 10 years from the date that the 2021 Plan was adopted. No ISO shall be exercisable after the expiration of 10 years after the effective date of grant of such award, subject to the following sentence. In the case of an ISO granted to a ten percent stockholder, (i) the exercise price shall not be less than 110% of the fair market value of a share on the date of grant of such ISO, and (ii) the exercise period shall not exceed 5 years from the effective date of grant of such ISO.
Restricted Stock and Restricted Stock Units. The committee may award restricted stock and RSUs under the 2021 Plan. Restricted stock awards consist of shares of stock that are transferred to the participant subject to restrictions that may result in forfeiture if specified vesting conditions are not satisfied. RSU awards result in the transfer of shares of stock to the participant only after specified vesting conditions are satisfied. A holder of restricted stock is treated as a current stockholder and shall be entitled to dividend and voting rights, whereas the holder of a restricted stock unit is treated as a stockholder with respect to the award only when the shares are delivered in the future. RSUs may include dividend equivalents. Specified vesting conditions may include performance goals to be achieved during any performance period and the length of the performance period. The committee may, in its discretion, make adjustments to performance goals based on certain changes in the Company’s business operations, corporate or capital structure or other circumstances. When the participant satisfies the conditions of an RSU award, the Company may settle the award (including any related dividend equivalent rights) in shares, cash or other property, as determined by the committee, in its sole discretion.
Other Shares or Share-Based Awards. The committee may grant other forms of equity-based or equity-related awards other than stock options, restricted stock or restricted stock units. The terms and conditions of each stock-based award shall be determined by the committee.
Clawback Rights. Awards granted under the 2021 Plan will be subject to recoupment or clawback under the Company’s clawback policy or applicable law, both as in effect from time to time.
Sale of the Company. Awards granted under the 2021 Plan automatically accelerate and vest, become exercisable (with respect to stock options), or have performance targets deemed earned at target level if there is a sale of the Company. The Company does not use a “liberal” definition of change in control as defined in Institutional Shareholder Services’ proxy voting guidelines.
No Repricing. The 2021 Plan prohibits the amendment of the terms of any outstanding award, and any other action taken in a manner to achieve (i) the reduction of the exercise price of NQSOs, ISOs or stock appreciation rights (collectively, “Stock Rights”); (ii) the cancellation of outstanding Stock Rights in exchange for cash or other awards with an exercise price that is less than the exercise price or base price of the original award; (iii) the cancellation of outstanding Stock Rights with an exercise price or base price that is less than the then current fair market value of a share of Common Stock in exchange for other awards, cash or other property; or (iv) otherwise effect a transaction that would be considered a “repricing” for the purposes of the stockholder approval rules of the applicable securities exchange or inter-dealer quotation system on which the Common Stock is listed or quoted without stockholder approval.
Transferability of Awards. Except as described below, awards under the 2021 Plan generally are not transferable by the recipient other than by will or the laws of descent and distribution. Any amounts payable or shares issuable pursuant to an award generally will be paid only to the recipient or the recipient’s beneficiary or representative. The committee has discretion, however, to permit certain transfer of awards to other persons or entities.
106
Adjustments. As is customary in incentive plans of this nature, each share limit and the number and kind of shares available under the 2021 Plan and any outstanding awards, as well as the exercise price or base price of awards, and performance targets under certain types of performance-based awards, are subject to adjustment in the event of certain reorganizations, mergers, combinations, recapitalizations, stock splits, stock dividends, or other similar events that change the number or kind of shares outstanding, and extraordinary dividends or distributions of property to the stockholders.
Amendment and Termination. The board of directors may amend, modify or terminate the 2021 Plan without stockholder approval, except that stockholder approval must be obtained for any amendment that, in the reasonable opinion of the board or the committee, constitute a material change requiring stockholder approval under applicable laws, policies or regulations or the applicable listing or other requirements of a stock exchange on which shares of Common Stock are then listed. The 2021 Plan will terminate upon the earliest of (1) termination of the 2021 Plan by the board of directors, or (2) the tenth anniversary of the board adoption of the 2021 Plan. Awards outstanding upon expiration of the 2021 Plan shall remain in effect until they have been exercised or terminated, or have expired.
107
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
In addition to the compensation arrangements, including employment, termination of employment and change in control arrangements and indemnification arrangements, discussed, when required, in the sections titled “Management” and “Executive Compensation,” the following is a description of each transaction since January 1, 2018 and each currently proposed transaction in which:
• we have been or are to be a participant;
• the amount involved exceeded or exceeds the lesser of $120,000 or 1% of our assets; and
• any of our directors, executive officers or holders of more than 5% of our capital stock, or any immediate family member of, or person sharing the household with, any of these individuals, had or will have a direct or indirect material interest.
Limitation of Liability and Indemnification of Officers and Directors
Prior to the completion of this offering, we expect to adopt an amended and restated certificate of incorporation, which will become effective immediately prior to the completion of this offering and which will contain provisions that limit the liability of our directors for monetary damages to the fullest extent permitted by Delaware law. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for the following:
• any breach of their duty of loyalty to our company or our stockholders;
• any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law;
• unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or
• any transaction from which they derived an improper personal benefit.
Any amendment to, or repeal of, these provisions will not eliminate or reduce the effect of these provisions in respect of any act, omission or claim that occurred or arose prior to that amendment or repeal. If the Delaware General Corporation Law is amended to provide for further limitations on the personal liability of directors of corporations, then the personal liability of our directors will be further limited to the greatest extent permitted by the Delaware General Corporation Law.
In addition, prior to the completion of this offering, we expect to adopt amended and restated bylaws which will provide that we will indemnify, to the fullest extent permitted by law, any person who is or was a party or is threatened to be made a party to any action, suit or proceeding by reason of the fact that he or she is or was one of our directors or officers or is or was serving at our request as a director or officer of another corporation, partnership, joint venture, trust or other enterprise. Our amended and restated bylaws are expected to provide that we may indemnify to the fullest extent permitted by law any person who is or was a party or is threatened to be made a party to any action, suit or proceeding by reason of the fact that he or she is or was one of our employees or agents or is or was serving at our request as an employee or agent of another corporation, partnership, joint venture, trust or other enterprise. Our amended and restated bylaws will also provide that we must advance expenses incurred by or on behalf of a director or officer in advance of the final disposition of any action or proceeding, subject to very limited exceptions.
Further, prior to the completion of this offering, we expect to enter into indemnification agreements with each of our directors and executive officers that may be broader than the specific indemnification provisions contained in the Delaware General Corporation Law. These indemnification agreements will require us, among other things, to indemnify our directors and executive officers against liabilities that may arise by reason of their status or service. These indemnification agreements will also require us to advance all expenses incurred by the directors and executive officers in investigating or defending any such action, suit or proceeding. We believe that these agreements are necessary to attract and retain qualified individuals to serve as directors and executive officers.
The limitation of liability and indemnification provisions that are expected to be included in our amended and restated certificate of incorporation, amended and restated bylaws and in indemnification agreements that we enter into with our directors and executive officers may discourage stockholders from bringing a lawsuit against our
108
directors and executive officers for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against our directors and executive officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be harmed to the extent that we pay the costs of settlement and damage awards against directors and executive officers as required by these indemnification provisions. At present, we are not aware of any pending litigation or proceeding involving any person who is or was one of our directors, officers, employees or other agents or is or was serving at our request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, for which indemnification is sought, and we are not aware of any threatened litigation that may result in claims for indemnification.
Prior to the completion of this offering, we expect to obtain insurance policies under which, subject to the limitations of the policies, coverage is provided to our directors and executive officers against loss arising from claims made by reason of breach of fiduciary duty or other wrongful acts as a director or executive officer, including claims relating to public securities matters, and to us with respect to payments that may be made by us to these directors and executive officers pursuant to our indemnification obligations or otherwise as a matter of law.
The underwriting agreement will provide for indemnification by the underwriters of us and our officers, directors and employees for certain liabilities arising under the Securities Act or otherwise. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling our company pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Policies and Procedures for Related Party Transactions
Following the completion of this offering, our audit committee charter will provide that the audit committee has the primary responsibility for reviewing and approving or disapproving “related party transactions,” which are transactions between us and related persons in which the aggregate amount involved exceeds or may be expected to exceed the lesser of $120,000 or 1% of our assets and in which a related person has or will have a direct or indirect material interest. For purposes of this policy, a related person will be defined as a director, executive officer, nominee for director or greater than 5% beneficial owner of our common stock, in each case since the beginning of the most recently completed year, and their immediate family members. As of the date of this prospectus, we have not adopted any formal standards, policies or procedures governing the review and approval of related party transactions, but we expect that our audit committee will do so in the future.
All of the transactions described above were entered into prior to the adoption of this policy. Accordingly, each was approved by disinterested members of our board of directors after making a determination that the transaction was executed on terms no less favorable than those that could have been obtained from an unrelated third party.
109
The following table sets forth information regarding the beneficial ownership of our common stock as of December 31, 2021 by (i) such persons known to us to be beneficial owners of more than 5% of our common stock, (ii) each of our directors and named executive officers, and (iii) all of our directors and executive officers as a group.
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to such securities. Beneficial ownership includes shares issuable pursuant to stock options that are exercisable within 60 days of December 31, 2021. The number of shares of our common stock beneficially owned and percentages of beneficial ownership before this offering that as set forth below are based on 15,069,930 shares of common stock outstanding, which includes 6,820,211 shares of our common stock outstanding as of December 31, 2021, plus 8,249,719 shares of our common stock issued upon the conversion of our preferred stock. The number of shares of our common stock outstanding and percentages of beneficial ownership after this offering that are set forth below includes 1,500,000 shares of common stock being offered for sale by us in this offering. The number of shares of our common stock beneficially owned and percentages of beneficial ownership do not reflect shares of common stock issuable upon conversion of a convertible debt agreement, dated September 20, 2021, with an aggregate principal plus accrued interest as of December 31, 2021 of $2,016,767, which will convert into shares of our common stock upon the closing of this offering.
To our knowledge, except as otherwise indicated, all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. Unless otherwise indicated, the address for each listed stockholder is: 61 Wilton Road, 3rd Floor Westport, CT 06880.
|
Name and Address of Beneficial Owner |
Common Stock |
Common Stock |
Common Stock |
|||||||||
|
Number |
Percentage |
Number |
Percentage |
Number |
Percentage |
|||||||
|
Directors and Named Executive Officers: |
||||||||||||
|
Lewis H. Bender(1) |
4,225,000 |
27.62 |
4,225,000 |
24.23 |
4,225,000 |
23.79 |
||||||
|
Dr. Ian B. Walters(2) |
616,250 |
3.97 |
616,250 |
3.49 |
616,250 |
3.42 |
||||||
|
Rebecca Drain(3) |
18,125 |
* |
18,125 |
* |
18,125 |
* |
||||||
|
Dr. Declan Doogan(4) |
146,495 |
* |
146,495 |
* |
146,495 |
* |
||||||
|
Dr. Emer Leahy(5) |
108,000 |
* |
108,000 |
* |
108,000 |
* |
||||||
|
Dr. Mark Goldberg(6) |
88,000 |
* |
88,000 |
* |
88,000 |
* |
||||||
|
All executive officers and directors as a group (8 persons) |
5,258,745 |
32.59 |
5,258,745 |
28.77 |
5,258,745 |
28.27 |
||||||
|
5% Stockholders: |
||||||||||||
|
Leonard Batterson(7) |
2,449,150 |
16.20 |
2,449,150 |
14.19 |
2,449,150 |
13.93 |
||||||
|
Larry Levy(8) |
1,446,749 |
9.58 |
1,446,749 |
8.39 |
1,446,749 |
8.23 |
||||||
|
Portage Biotech Inc.(9) |
1,288,458 |
8.55 |
1,288,458 |
7.49 |
1,288,458 |
7.35 |
||||||
|
Craig J. Duchossois(10) |
1,265,620 |
8.38 |
1,265,620 |
7.34 |
1,265,620 |
7.21 |
||||||
____________
* Less than 1 percent
(1) Consists of (i) 4,000,000 shares of common stock and (ii) 225,000 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
(2) Consists of (i) 150,000 shares of common stock and (ii) 466,250 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
(3) Consists of 18,125 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
110
(4) Consists of (i) 25,000 shares of common stock issuable upon conversion of Series A Preferred Stock, (ii) 4,614 shares of common stock issuable upon conversion of Series B Preferred Stock, (iii) 3,881 shares of common stock issuable upon conversion of Series C Preferred Stock and (iv) 113,000 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
(5) Consists of 108,000 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
(6) Consists of 88,000 shares of common stock issuable upon the exercise of options exercisable within 60 days after October 1, 2021.
(7) Consists of (i) 346,000 shares of common stock held by VCapital Intensity LLC, (ii) 814,833 shares of common stock issuable upon conversion of Series A Preferred Stock held by BVC — Intensity LLC, (iii) 381,111 shares of common stock issuable upon conversion of Series B Preferred Stock held by VCapital Intensity LLC, (iv) 822,423 shares of common stock issuable upon conversion of Series C Preferred Stock held by VCapital Intensity LLC, (v) 34,783 shares of common stock issuable upon conversion of Series C Preferred Stock held by BVC — Intensity LLC, and (vi) 50,000 shares of common stock issuable upon the exercise of warrants exercisable within 60 days after October 1, 2021. Does not include shares of common stock issuable upon conversion of a convertible debt agreement, dated September 20, 2021, held by VCapital Intensity LLC with an aggregate principal of $2,000,000, which will convert into shares of our common stock upon the closing of this offering based on the unpaid principal and accrued interest on such date. Mr. Batterson may be deemed to beneficially own such shares. The principal business address of VCapital Intensity LLC and BVC — Intensity LLC is 901 W. Jackson Blvd., Suite 503 Chicago, IL 60607.
(8) Consists of (i) 775,000 shares of common stock issuable upon conversion of Series A Preferred Stock held by LFP River West Investors, LLC — Series 21, (ii) 244,445 shares of common stock issuable upon conversion of Series B Preferred Stock held by LFP River West Investors, LLC — Series 38, (iii) 391,304 shares of common stock issuable upon conversion of Series C Preferred Stock held by LFP River West Investors, LLC — Series 38 and (iv) 36,600 shares of common stock issuable upon the exercise of warrants exercisable within 60 days after October 1, 2021. Mr. Levy may be deemed to beneficially own such shares. The registered address for LFP River West Investors, LLC is 251 Little Falls Drive, Wilmington, DE 19808.
(9) Consists of (i) 1,250,000 shares of common stock issuable upon conversion of Series A Preferred Stock and (ii) 38,458 shares of common stock issuable upon conversion of Series B Preferred Stock. Portage Biotech Inc., a publicly traded company incorporated under the laws of the British Virgin Islands with disparate ownership, is governed by a board of directors, including Dr. Doogan who is one of our directors, and is managed by its executive officers; accordingly, no natural persons control Portage Biotech Inc. The principal business address of Portage Biotech Inc. is Craigmuir Chambers, Road Town, Tortola, British Virgin Islands, VG1110.
(10) Consists of (i) 750,000 shares of common stock issuable upon conversion of Series A Preferred Stock, (ii) 230,750 shares of common stock issuable upon conversion of Series B Preferred Stock and (iii) 260,870 shares of common stock issuable upon conversion of Series C Preferred Stock and (iv) 24,000 shares of common stock issuable upon the exercise of warrants exercisable within 60 days after October 1, 2021. All shares are held by Craig J. Duchossois Revocable Trust UAD 9/11/1989. Mr. Duchossois may be deemed to beneficially own such shares. The principal business address of Craig J. Duchossois is 444 W. Lake St, Suite 2000, Chicago, Illinois 60606.
111
General
The following description of our capital stock and certain provisions of our amended and restated certificate of incorporation and amended and restated bylaws are summaries and are qualified by reference to our amended and restated certificate of incorporation and our amended and restated bylaws, which will become effective immediately prior to the completion of this offering. Copies of these documents will be filed with the SEC as exhibits to our registration statement, of which this prospectus forms a part. The descriptions of the common stock and preferred stock reflect changes to our capital structure that will be in effect immediately prior to the completion of this offering.
Upon filing of our amended and restated certificate of incorporation and the closing of this offering, our authorized capital stock will consist of 150,000,000 shares, all with a par value of $0.0001 per share, of which 135,000,000 shares will be designated common stock and 15,000,000 shares will be designated preferred stock.
As of immediately before the completion of this offering, after giving effect to the conversion of our preferred stock into 8,249,719 shares of our common stock and the conversion of a convertible note into an aggregate of 381,265 shares of our common stock (which is based on unpaid principal and accrued but unpaid interest as of September 30, 2021 at a conversion price of $5.25 per share), there will be 15,451,195 shares of common stock outstanding and held of record by 78 stockholders.
Common Stock
Voting Rights. The common stock is entitled to one vote per share on any matter that is submitted to a vote of our stockholders. Our amended and restated certificate of incorporation does not provide for cumulative voting for the election of directors. Our amended and restated certificate of incorporation establishes a classified board of directors that is divided into three classes with staggered three-year terms. Only the directors in one class will be subject to election by a plurality of the votes cast at each annual meeting of our stockholders, with the directors in the other classes continuing for the remainder of their respective three-year terms. The affirmative vote of holders of at least 66 2/3% of the voting power of all of the then outstanding shares of capital stock, voting as a single class, will be required to amend certain provisions of our amended and restated certificate of incorporation, including provisions relating to amending our amended and restated bylaws, the classified structure of our board of directors, the size of our board of directors, removal of directors, director liability, vacancies on our board of directors, special meetings, stockholder notices, actions by written consent and exclusive jurisdiction.
Dividends. Subject to preferences that may apply to any shares of preferred stock outstanding at the time, the holders of our common stock are entitled to receive dividends out of funds legally available if our board of directors, in its discretion, determines to issue dividends and then only at the times and in the amounts that our board of directors may determine. See the section titled “Dividend Policy” for further information.
Liquidation Rights. On our liquidation, dissolution, or winding-up, the holders of common stock will be entitled to share equally, identically and ratably in all assets remaining after the payment of any liabilities, liquidation preferences and accrued or declared but unpaid dividends, if any, with respect to any outstanding preferred stock, unless a different treatment is approved by the affirmative vote of the holders of a majority of the outstanding shares of such affected class, voting separately as a class.
No Preemptive or Similar Rights. The holders of our shares of common stock are not entitled to preemptive rights, and are not subject to conversion, redemption or sinking fund provisions.
Preferred Stock
Under our amended and restated certificate of incorporation, our board of directors may, without further action by our stockholders, fix the rights, preferences, privileges and restrictions of up to an aggregate of 15,000,000 shares of preferred stock in one or more series and authorize their issuance. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of our common stock. Any issuance of our preferred stock could adversely affect the voting power of holders of our common stock, and the likelihood that such holders would receive dividend payments and payments on liquidation. In
112
addition, the issuance of preferred stock could have the effect of delaying, deferring or preventing a change of control or other corporate action. Immediately prior to the completion of this offering, no shares of preferred stock will be outstanding. We have no present plan to issue any shares of preferred stock.
Anti-Takeover Provisions
Certificate of Incorporation and Bylaws to be in Effect Immediately Prior to the Completion of this Offering.
Because our stockholders do not have cumulative voting rights, stockholders holding a majority of the voting power of our shares of common stock will be able to elect all our directors. Our amended and restated certificate of incorporation and our amended and restated bylaws will require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent or electronic transmission. A special meeting of stockholders may be called by a majority of our board of directors, the chair of our board of directors, our chief executive officer or our lead independent director. Our amended and restated bylaws will establish an advance notice procedure for stockholder proposals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors.
As described above in “Management — Board Composition and Election of Directors,” in accordance with our amended and restated certificate of incorporation to be filed in connection with this offering, immediately after this offering, our board of directors will be divided into three classes with staggered three-year terms.
The foregoing provisions will make it more difficult for another party to obtain control of us by replacing our board of directors. Since our board of directors has the power to retain and discharge our officers, these provisions could also make it more difficult for existing stockholders or another party to effect a change in management. In addition, the authorization of undesignated preferred stock makes it possible for our board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change our control.
These provisions are designed to reduce our vulnerability to an unsolicited acquisition proposal and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and may have the effect of deterring hostile takeovers or delaying changes in our control or management. As a consequence, these provisions may also inhibit fluctuations in the market price of our stock that could result from actual or rumored takeover attempts.
Section 203 of the Delaware General Corporation Law
When we have a class of voting stock that is either listed on a national securities exchange or held of record by more than 2,000 stockholders, we will be subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, subject to certain exceptions.
Choice of Forum
Our amended and restated certificate of incorporation to be effective immediately prior to the completion of this offering will provide that unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware (or, if and only if the Court of Chancery of the State of Delaware lacks subject matter jurisdiction, any state court located within the State of Delaware or, if and only if all such state courts lack subject matter jurisdiction, the federal district court for the District of Delaware) and any appellate court therefrom shall be the sole and exclusive forum for the following claims or causes of action under Delaware statutory or common law: (A) any derivative claim or cause of action brought on our behalf; (B) any claim or cause of action for breach of a fiduciary duty owed by any of our current or former directors, officers or other employees to us or our stockholders; (C) any claim or cause of action against us or any of our current or former directors, officers or other employees arising out of or pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation or our amended and restated bylaws (as each may be amended from time to time); (D) any claim or cause of action seeking to interpret, apply, enforce or determine the validity of our amended and restated certificate of incorporation or our amended and restated bylaws (as each may be amended from time to time, including any right, obligation or remedy thereunder); (E) any claim or cause of action as to which the Delaware General Corporation Law confers jurisdiction on the Court of Chancery of the State of Delaware; and (F) any claim or cause of action against us or any of our current or former directors, officers or other employees governed by the internal-affairs doctrine or
113
otherwise related to our internal affairs, in all cases to the fullest extent permitted by law and subject to the court having personal jurisdiction over the indispensable parties named as defendants; provided, that, this Delaware forum provision set forth in our amended and restated certificate of incorporation to be effective immediately prior to the completion of this offering shall not apply to claims or causes of action brought to enforce a duty or liability created by the Securities Act or the Exchange Act, or any other claim for which the federal courts have exclusive jurisdiction.
Further, unless we consent in writing to the selection of an alternative forum, to the fullest extent permitted by law, the federal district courts of the United States shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act, including all causes of action asserted against any defendant named in such complaint. For the avoidance of doubt, these provisions are intended to benefit and may be enforced by us, our officers and directors, the underwriters for any offering giving rise to such complaint, and any other professional entity whose profession gives authority to a statement made by that person or entity and who has prepared or certified any part of the documents underlying the offering. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions, and there can be no assurance that such provisions will be enforced by a court in those other jurisdictions. We note that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder.
Limitations of Liability and Indemnification
See “Certain Relationships and Related Party Transactions — Limitation of Liability and Indemnification of Officers and Directors.”
Exchange Listing
We have applied to list our common stock on the Nasdaq Capital Market under the symbol “INTS.”
Transfer Agent and Registrar
Upon the completion of this offering, the transfer agent and registrar for our common stock will be Continental Stock Transfer & Trust Company. The transfer agent’s address is 1 State Street, 30th Floor, New York, NY 10004-1561.
114
SHARES ELIGIBLE FOR FUTURE SALE
Immediately prior to this offering, there was no public market for our common stock, and we cannot predict what effect, if any, market sales of shares of common stock or the availability of shares of common stock for sale will have on the market price of our common stock prevailing from time to time. Nevertheless, sales of substantial amounts of common stock, including shares issued upon the exercise of outstanding options and warrants, in the public market, or the perception that such sales could occur, could materially and adversely affect the market price of our common stock and could impair our future ability to raise capital through the sale of our equity or equity-related securities at a time and price that we deem appropriate.
Upon the completion of this offering, we will have outstanding an aggregate of approximately 17,594,053 shares of common stock (or 17,915,481 shares of common stock if the underwriters’ option to purchase additional shares of common stock is exercised in full). In addition, options and warrants to purchase an aggregate of approximately 2,469,000 shares of our common stock will be outstanding as of the completion of this offering. Of the outstanding shares, the shares sold in this offering will be freely tradable without restriction or further registration under the Securities Act, except any shares purchased by our “affiliates,” as that term is defined in Rule 144 under the Securities Act, may be sold only in compliance with the limitations described below. The remaining outstanding shares of common stock will be deemed restricted securities, as defined under Rule 144. Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration under Rules 144 or 701 under the Securities Act, which we summarize below. All of these shares will be subject to lock-up agreements described below.
Taking into account the lock-up agreements described below, and assuming A.G.P./Alliance Global Partners, as representative of the underwriters, does not release stockholders from these agreements, certain shares will be eligible for sale in the public market at the following times, subject to the provisions of Rule 144 and Rule 701.
Rule 144
In general, under Rule 144 as currently in effect, once we have been subject to public company reporting requirements for at least 90 days, a person who is not deemed to have been one of our affiliates for purposes of the Securities Act at any time during the 90 days preceding a sale and who has beneficially owned the shares proposed to be sold for at least six months, including the holding period of any prior owner other than our affiliates, is entitled to sell such shares (subject to the requirements of the lock-up agreements, as described below) without complying with the manner of sale, volume limitation or notice provisions of Rule 144, subject to compliance with the public information requirements of Rule 144. If such a person has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any prior owner other than our affiliates, then such person is entitled to sell such shares (subject to the requirements of the lock-up agreements, as described below) without complying with any of the requirements of Rule 144.
In general, under Rule 144, as currently in effect, our affiliates or persons selling shares on behalf of our affiliates are entitled to sell upon expiration of the lock-up agreements described below, within any three-month period beginning 90 days after the date of this prospectus, a number of shares that does not exceed the greater of one percent of the number of shares of our common stock then outstanding or the average weekly trading volume of our common stock on Nasdaq during the four calendar weeks preceding the date of filing of a Notice of Proposed Sale of Securities Pursuant to Rule 144 with respect to the sale:
Sales under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us. Notwithstanding the availability of Rule 144, other stockholders owning an aggregate of approximately 11,975,607 shares of our common stock (on a pro forma basis) have entered into lock-up agreements as described below, and their restricted securities will become eligible for sale (subject to the above limitations under Rule 144) upon the expiration of the restrictions set forth in those agreements.
Rule 701
In general, under Rule 701 as currently in effect, any of our employees, directors, officers, consultants or advisors who purchase shares from us in connection with a compensatory stock or option plan or other written agreement before the completion of this offering is entitled to sell such shares (subject to the requirements of the lock-up agreements, as
115
described below) 90 days after the completion of this offering in reliance on Rule 144, in the case of affiliates, without having to comply with the holding period requirements of Rule 144 and, in the case of non-affiliates, without having to comply with the public information, holding period, volume limitation or notice filing requirements of Rule 144.
Lock-Up Agreements
Notwithstanding the availability of Rule 144, we and all of our officers, directors and four of our stockholders owning an aggregate of approximately 11,975,607 shares of our common stock (on a pro forma basis), or securities exercisable for or convertible into our common stock outstanding immediately prior to this offering, have agreed that, without the prior written consent of A.G.P./Alliance Global Partners, as representative of the underwriters, we and they will not, during the period ending 180 days after the date of this prospectus:
• offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend or otherwise transfer or dispose of, directly or indirectly, any shares of common stock or any securities convertible into or exercisable or exchangeable for shares of common stock; or
• enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of our common stock, whether any such transaction described above is to be settled by delivery of shares of our common stock or such other securities, in cash or otherwise, subject to certain exceptions set forth in the section entitled “Underwriting.”
Registration Statements on Form S-8
We intend to file one or more registration statements on Form S-8 under the Securities Act with the SEC to register the offer and sale of shares of our common stock that are issuable under our 2013 Stock and Option Plan, or the 2013 Plan, and our proposed 2021 Stock Incentive Plan, or the 2021 Plan. These registration statements will become effective immediately on filing. Shares covered by these registration statements will then be eligible for sale in the public markets, subject to vesting restrictions, any applicable lock-up agreements and market standoff provisions described below, and Rule 144 limitations applicable to affiliates.
116
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS
FOR NON-U.S. HOLDERS OF COMMON STOCK
The following is a summary of certain material U.S. federal income tax considerations relating to the acquisition, ownership and disposition of shares of our common stock issued pursuant to this offering by “non-U.S. holders,” as defined below. This summary deals only with shares of our common stock acquired by a non-U.S. holder in this offering that are held as capital assets within the meaning of Section 1221 of the Internal Revenue Code of 1986, as amended (the “Code”) (generally, property held for investment). This summary does not address all aspects of U.S. federal income taxation that may be important to a particular non-U.S. holder in light of that non-U.S. holder’s individual circumstances, nor does it address any aspects of the unearned income Medicare contribution tax pursuant to the Health Care and Education Reconciliation Act of 2010, any U.S. federal gift and estate taxes, except to the limited extent provided below, any U.S. alternative minimum taxes or any state, local or non-U.S. taxes. This summary does not address the U.S. federal income tax considerations applicable to a non-U.S. holder that is subject to special treatment under U.S. federal income tax laws, including: a broker or dealer in securities or currencies; a financial institution; a tax-exempt organization (including a private foundation) and a tax-qualified retirement plan; a non-U.S. government or an international organization; a “qualified foreign pension fund” as defined in Section 897(l)(2) of the Code and an entity all of the interests of which are held by qualified foreign pension funds; an insurance company; a person holding shares of our common stock as part of a hedging, integrated, conversion or straddle transaction or a person deemed to sell shares of our common stock under the constructive sale provisions of the Code; a trader in securities that has elected the mark-to-market method of accounting; an entity or arrangement that is treated as a partnership (or is disregarded from its owner) for U.S. federal income tax purposes; a person that received shares of our common stock in connection with services provided to us or any of our affiliates; a person subject to special tax accounting rules as a result of any item of gross income with respect to our common stock being taken into account in an applicable consolidated financial statement; a person that owns, or is deemed to own, more than five percent of our common stock; a person whose “functional currency” is not the U.S. dollar; a “controlled foreign corporation”; a “passive foreign investment” company; a corporation that accumulates earnings to avoid U.S. federal income tax; and U.S. expatriates and certain former citizens or long-term residents of the United States.
This summary is based upon provisions of the Code, and applicable Treasury regulations promulgated or proposed thereunder, rulings and judicial decisions, all as in effect as of the date hereof. Those authorities may be changed, perhaps with retroactive effect, or may be subject to differing interpretations, which could result in U.S. federal income tax consequences different from those discussed below. There can be no assurance that the Internal Revenue Service (“IRS”) will concur with the discussion of the tax considerations set forth below, and we have not obtained, and we do not intend to obtain, a ruling from the IRS with respect to the U.S. federal income tax consequences to a non-U.S. holder of the purchase, ownership or disposition of shares of our common stock. This summary does not address all aspects of U.S. federal income tax and does not address any state, local, non-U.S., or gift tax considerations or any considerations relating to the alternative minimum tax or the Medicare tax on net investment income.
For purposes of this discussion, a “non-U.S. holder” is a beneficial holder of shares of our common stock that is for U.S. federal income tax purposes not a partnership or disregarded entity and not (i) an individual citizen or resident of the United States for U.S. federal income tax purposes; (ii) a corporation (or any other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof or the District of Columbia (or otherwise treated as a domestic corporation for U.S. federal income tax purposes); (iii) an estate the income of which is subject to U.S. federal income taxation regardless of its source; or (iv) a trust if it (1) is subject to the primary supervision of a court within the United States and one or more U.S. persons (as defined in the Code) have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person.
An individual non-U.S. citizen may, in some cases, be deemed to be a resident alien (as opposed to a nonresident alien) by virtue of being present in the United States for at least 31 days in the calendar year and for an aggregate of at least 183 days during a three-year period ending in the current calendar year. Generally, for this purpose, all the days present in the current year, one-third of the days present in the immediately preceding year, and one-sixth of the days present in the second preceding year, are counted.
117
Resident aliens are generally subject to U.S. federal income tax as if they were U.S. citizens. Individuals who are uncertain of their status as resident or nonresident aliens for U.S. federal income tax purposes are urged to consult their tax advisors regarding the U.S. federal income tax consequences of the ownership or disposition of our common stock.
If an entity or arrangement that is treated as a partnership for U.S. federal income tax purposes holds shares of our common stock, the tax treatment of a person treated as a partner in such partnership for U.S. federal income tax purposes generally will depend upon the status of the partner and the activities of the partnership. Any entity or arrangement that is treated as a partnership for U.S. federal income tax purposes, and any person holding shares of our common stock through such a partnership, are urged to consult their tax advisors regarding the acquisition, ownership and disposition of shares of our common stock.
This summary is for general information only and is not, and is not intended to be, tax advice. Non-U.S. holders of shares of our common stock are urged to consult their tax advisors concerning the tax considerations related to the acquisition, ownership and disposition of shares of our common stock in light of their particular circumstances, as well as any tax considerations relating to gift or estate taxes, the alternative minimum tax or to the Medicare tax on net investment income, and any tax considerations arising under the laws of any other jurisdiction, including any state, local and non-U.S. income and other tax laws or under any applicable tax treaty.
Distributions
As discussed in the section entitled “Dividend Policy” above, we do not currently expect to make distributions in respect of our common stock. In the event that we do make a distribution of cash or property with respect to our common stock, any such distributions generally will constitute dividends for U.S. federal income tax purposes to the extent of our current and accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will first constitute a return of capital and will reduce a holder’s adjusted tax basis in such holder’s shares of our common stock, determined on a share-per-share basis but not below zero. Any remaining excess will be treated as capital gain and subject to the tax treatment described below in the section entitled “— Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock.”
Unless dividends, if any, are effectively connected with a non-U.S. holder’s U.S. trade or business (and if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base maintained in the United States), dividends paid to a non-U.S. holder of shares of our common stock generally will be subject to U.S. federal income tax (which generally will be collected through withholding) at a rate of 30% of the gross amount of the dividends (or such lower rate specified by an applicable income tax treaty). Even if a non-U.S. holder is eligible for a lower treaty rate, dividend payments generally will be subject to withholding at a 30% rate (rather than the lower treaty rate) unless the non-U.S. holder provides a valid IRS Form W-8BEN or W-8BEN-E or other appropriate form (or any successor or substitute form thereof) certifying such holder’s qualification for the reduced rate. Such form must be provided prior to the payment of the applicable dividend and must be updated periodically. If a non-U.S. holder holds stock through a financial institution or other agent acting on the holder’s behalf, the holder will be required to provide appropriate documentation to such agent. The holder’s agent will then be required to provide certification to the applicable withholding agent, either directly or through other intermediaries. Each non-U.S. holder should consult its tax advisor regarding its entitlement to benefits under an applicable income tax treaty.
Subject to the discussions below regarding backup withholding and the Foreign Account Tax Compliance Act, if dividends paid to a non-U.S. holder are effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States (and, if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base maintained in the United States), the non-U.S. holder will be exempt from U.S. federal withholding tax. To claim the exemption, the non-U.S. holder must furnish to us or the relevant withholding agent a valid IRS Form W-8ECI or other appropriate form (or any successor or substitute form thereof), certifying that the dividends are effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States.
Any dividends paid on shares of our common stock that are effectively connected with a non-U.S. holder’s U.S. trade or business (and, if required by an applicable tax treaty, attributable to a permanent establishment or fixed base maintained in the United States) generally will be subject to U.S. federal income tax on a net income basis in the same manner as if such holder were a U.S. person. A non-U.S. holder that is a corporation also may be subject to a branch
118
profits tax at a rate of 30% (or such lower rate specified by an applicable tax treaty) on a portion of its effectively connected earnings and profits for the taxable year. Non-U.S. holders should consult their tax advisors regarding any applicable tax treaties that may provide for different rules.
Non-U.S. holders who do not timely provide us or the relevant withholding agent with the required certification, but who qualify for a reduced treaty rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non-U.S. holders should consult their tax advisors regarding their entitlement to benefits under a tax treaty.
If at the time a distribution is made we are not able to determine whether or not it will be treated as a dividend for U.S. federal income tax purposes (as opposed to being treated as a return of capital or capital gain), we or a financial intermediary may withhold tax on all or a portion of such distribution at the rate applicable to dividends. However, a non-U.S. holder may obtain a refund of any excess withholding by timely filing an appropriate claim for refund with the IRS.
Any distribution described in this section would also be subject to the discussion below in the section entitled “Foreign Account Tax Compliance Act.”
Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock
Subject to the discussions below regarding backup withholding and the Foreign Account Tax Compliance Act, a non-U.S. holder generally will not be subject to U.S. federal income tax or withholding tax on gain realized upon a sale, exchange or other taxable disposition of shares of our common stock (including a redemption, but only if the redemption would be treated as a sale or exchange rather than as a distribution for U.S. federal income tax purposes) unless: (i) the gain is effectively connected with the non-U.S. holder’s conduct of a trade or business in the United States (and, if required by an applicable income tax treaty, is attributable to a permanent establishment or fixed base maintained in the United States); (ii) the non-U.S. holder is a non-resident alien individual who is present in the United States for 183 days or more in the taxable year of that disposition, and certain other conditions are met; or (iii) we are or have been a “U.S. real property holding corporation” (“USRPHC”) for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding the disposition and the non-U.S. holder’s holding period for shares of our common stock (the “relevant period”) and certain other conditions are met, as described below.
If the first exception applies, the non-U.S. holder generally will be subject to U.S. federal income tax on a net basis with respect to such gain in the same manner as if such holder were a resident of the United States. In addition, if the non-U.S. holder is a corporation for U.S. federal income tax purposes, such gains may, under certain circumstances, also be subject to the branch profits tax at a rate of 30% (or at a lower rate prescribed by an applicable income tax treaty).
If the second exception applies, the non-U.S. holder generally will be subject to U.S. federal income tax at a rate of 30% on the gain from a disposition of shares of our common stock, which may be offset by capital losses allocable to U.S. sources during the taxable year of disposition (even though the non-U.S. holder is not considered a resident of the United States), provided such holder timely filed U.S. federal income tax returns with respect to such losses.
With respect to the third exception above, we believe we currently are not, and we do not anticipate becoming, a USRPHC for U.S. federal income tax purposes. Because the determination of whether we are a USRPHC depends on the fair market value of our U.S. real property interests relative to the fair market value of our other trade or business assets and our non-U.S. real property interests, there can be no assurances that we will not become a USRPHC in the future. Generally, a corporation is a USRPHC only if the fair market value of its U.S. real property interests (as defined in the Code) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. Even if we are or become a USRPHC, a non-U.S. holder would not be subject to U.S. federal income tax on a sale, exchange or other taxable disposition of shares of our common stock by reason of our status as a USRPHC so long as (i) shares of our common stock continue to be regularly traded on an established securities market (within the meaning of Section 897(c)(3) of the Code) during the calendar year in which such disposition occurs and (ii) such non-U.S. holder does not own and is not deemed to own (directly, indirectly or constructively) more than 5% of the shares of our common stock at any time during the relevant period. If we are a USRPHC and the requirements described in clauses (i) or (ii) in the preceding sentence are not met, gain on the disposition of shares of our common stock generally will be taxed in the same manner as gain that is effectively
119
connected with the conduct of a U.S. trade or business, except that the branch profits tax generally will not apply. No assurance can be provided that our common stock will be regularly traded on an established securities market at all times for purposes of the rules described above.
Non-U.S. holders should consult their tax advisors regarding any applicable tax treaties that may provide for different rules.
Information Reporting and Backup Withholding Tax
We or a financial intermediary must report annually to the IRS and to each non-U.S. holder the gross amount of the distributions on shares of our common stock paid to such holder and the tax withheld, if any, with respect to such distributions, regardless of whether withholding was required. This information also may be made available under a specific treaty or agreement with the tax authorities in the country in which the non-U.S. holder resides or is established. A non-U.S. holder generally will be subject to backup withholding at the then applicable rate for dividends paid to such holder unless such holder furnishes a valid IRS Form W-8BEN or W-8BEN-E (or such other applicable form and documentation as required by the Code or the Treasury regulations) certifying under penalties of perjury that it is a non-U.S. holder (and the payor does not have actual knowledge or reason to know that such holder is a U.S. person as defined under the Code), or otherwise establishes an exemption. Dividends paid to non-U.S. holders subject to U.S. federal withholding tax, as described above in the section entitled “Distributions,” generally will be exempt from U.S. backup withholding.
Information reporting and, depending on the circumstances, backup withholding will apply to the payment of the proceeds of a sale or other disposition of shares of our common stock by a non-U.S. holder effected by or through the U.S. office of any broker, U.S. or non-U.S., unless such holder certifies that it is not a U.S. person (as defined under the Code) and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup withholding will not apply to a payment of disposition proceeds to a non-U.S. holder where the transaction is effected outside the U.S. through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker. Prospective investors should consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a credit against a non-U.S. holder’s U.S. federal income tax liability, if any, and may entitle such holder to a refund, provided that an appropriate claim is timely filed with the IRS.
Foreign Account Tax Compliance Act
Under legislation commonly referred to as the Foreign Account Tax Compliance Act, as modified by Treasury regulations and subject to any official interpretations thereof, any applicable intergovernmental agreement between the United States and a non-U.S. government to implement these rules and improve international tax compliance, or any fiscal or regulatory legislation or rules adopted pursuant to any such agreement (collectively, “FATCA”), a 30% withholding tax will apply to dividends, if any, on, and, subject to the proposed Treasury Regulations discussed below, gross proceeds from the sale or other disposition of, shares of our common stock paid to certain non-U.S. entities (including financial intermediaries) unless various information reporting and due diligence requirements, which are different from and in addition to the certification requirements described elsewhere in this discussion, have been satisfied (generally relating to ownership by U.S. persons of interests in or accounts with those entities).
While, beginning on January 1, 2019, withholding under FATCA would have applied also to payments of gross proceeds from the sale or other disposition of our common stock, proposed Treasury Regulations eliminate FATCA withholding on payments of gross proceeds entirely. Taxpayers generally may rely on these proposed Treasury Regulations until final Treasury Regulations are issued.
Holders of shares of our common stock should consult their tax advisors regarding the possible impact of FATCA on their investment in our common stock, including, without limitation, the process and deadlines for meeting the applicable requirements to prevent the imposition of the 30% withholding tax under FATCA.
120
Federal Estate Tax
Common stock we have issued that is owned (or treated as owned) by an individual who is not a citizen or a resident of the United States (as defined for U.S. federal estate tax purposes) at the time of death will be included in the individual’s gross estate for U.S. federal estate tax purposes unless an applicable estate or other tax treaty provides otherwise, and therefore may be subject to U.S. federal estate tax. Holders of our common stock are urged to consult their tax advisors regarding the U.S. federal estate tax consequences of the ownership or disposition of our common stock.
Each prospective investor should consult its tax advisor regarding the particular U.S. federal, state, local, and non-U.S. tax consequences of purchasing, holding, and disposing of our common stock, including the consequences of any proposed change in applicable laws.
121
A.G.P./Alliance Global Partners (“AGP”) is acting as the sole book-running manager of the offering and as representative of the underwriters named below. Subject to the terms and conditions of the underwriting agreement dated the date of this prospectus, the underwriters named below, through the representative, have severally agreed to purchase, and we have agreed to sell to the underwriters, the following respective number of shares of Common Stock set forth opposite the underwriter’s name below:
|
Underwriter |
Number of |
|
|
A.G.P./Alliance Global Partners Corp. |
||
|
Brookline Capital Markets, a division of Arcadia Securities, LLC |
|
|
|
Total |
|
The underwriting agreement provides that the obligation of the underwriters to purchase the shares of Common Stock offered by this prospectus is subject to certain conditions. The underwriters are obligated to purchase all of the shares of Common Stock (other than those covered by the over-allotment option to purchase additional shares of Common Stock described below) offered hereby if any of the shares are purchased.
Underwriting Discounts, Commissions and Expenses
We have agreed to sell the securities to the underwriters at the offering price of $ per share of common stock, which represents the offering price of such securities set forth on the cover page of this prospectus, less the applicable 7% underwriting discount.
We have also agreed to reimburse the underwriters for accountable legal expenses not to exceed $75,000 and non-accountable expenses not to exceed 1% of the aggregate gross proceeds of this offering. We estimate that expenses payable by us in connection with this offering, including reimbursement of the underwriters’ out-of-pocket expenses, but excluding the underwriting discount referred to above, will be approximately $900,000.
The following table shows the underwriting discounts and commissions payable to the underwriters by us in connection with this offering (assuming both the exercise and non-exercise of the over-allotment option to purchase additional shares of Common Stock we have granted to the underwriters).
|
Paid by the Company |
||||||||||||
|
No Exercise of |
Full Exercise of |
|||||||||||
|
Per Share |
Total |
Per Share |
Total |
|||||||||
|
Public Offering Price |
$ |
$ |
$ |
$ |
||||||||
|
Underwriting discounts and commissions (7%) |
|
|
|
|
||||||||
|
Proceeds to us, before expenses |
$ |
$ |
$ |
$ |
||||||||
Over-Allotment Option to Purchase Additional Shares
Pursuant to the underwriting agreement, we have granted the underwriters an option, exercisable for up to 45 days from the date of this prospectus, to purchase up to additional shares of common stock on the same terms as the other shares of common stock being purchased by the underwriters from us. The underwriters may exercise the option solely to cover over-allotments. If the over-allotment option to purchase additional shares of Common Stock is exercised in full, the total public offering price, underwriting compensation (including discounts, but not including any other compensation described hereunder) and proceeds to us before offering expenses will be approximately $ million.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act and liabilities arising from breaches of representations and warranties contained in the underwriting agreement, or to contribute to payments that the underwriters may be required to make in respect any of those liabilities.
122
Lock-Up Agreements
In connection with this offering, we, along with our directors and executive officers have agreed with AGP, as representative of the underwriters, that for a 180-day “lock-up” period, commencing from the date of this prospectus, subject to specified exceptions, without the prior written consent of AGP, as representative of the underwriters, we and they will not offer, sell, pledge or otherwise dispose of these securities.
Price Stabilization, Short Positions, and Penalty Bids
The underwriters have advised us that they do not intend to conduct any stabilization or over-allotment activities in connection with this offering.
Passive Market Making
In connection with this offering, the underwriters and any selling group members may engage in passive market making transactions in our common stock on Nasdaq in accordance with Rule 103 of Regulation M under the Securities Exchange Act of 1934, as amended, during a period before the commencement of offers or sales of common stock and extending through the completion of the distribution. A passive market maker must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered below the passive market maker’s bid that bid must then be lowered when specified purchase limits are exceeded.
Electronic Distribution
This prospectus in electronic format may be made available on websites or through other online services maintained by the underwriters, or by their affiliates. Other than this prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by any of the underwriters is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriters in their capacity as underwriter, and should not be relied upon by investors.
Other
From time to time, the underwriters and/or its affiliates have provided, and may in the future provide, various investment banking and other financial services for us which services they have received and may in the future receive, customary fees. In the course of their businesses, the underwriters and their affiliates may actively trade our securities or loans for its own account or for the accounts of customers, and, accordingly, the underwriters and their affiliates may at any time hold long or short positions in such securities or loans. Except for services provided in connection with this offering, the underwriters have not provided any investment banking or other financial services to us during the 180-day period preceding the date of this prospectus, and we do not expect to retain the underwriters to perform any investment banking or other financial services for at least 90 days after the date of this prospectus.
Selling Restrictions
This prospectus does not constitute an offer to sell to, or a solicitation of an offer to buy from, anyone in any country or jurisdiction (i) in which such an offer or solicitation is not authorized, (ii) in which any person making such offer or solicitation is not qualified to do so or (iii) in which any such offer or solicitation would otherwise be unlawful. No action has been taken that would, or is intended to, permit a public offer of the securities or possession or distribution of this prospectus or any other offering or publicity material relating to the securities in any country or jurisdiction (other than the United States) where any such action for that purpose is required. Accordingly, the underwriters have undertaken that they will not, directly or indirectly, offer or sell any securities or have in its possession, distribute or publish any prospectus, form of application, advertisement or other document or information in any country or jurisdiction except under circumstances that will, to the best of its knowledge and belief, result in compliance with any applicable laws and regulations and all offers and sales of securities by it will be made on the same terms.
123
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any securities may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any securities may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
• to legal entities which are qualified investors as defined under the Prospectus Directive;
• by the underwriters to fewer than 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives of the underwriters for any such offer; or
• in any other circumstances falling within Article 3(2) of the Prospectus Directive,
provided that no such offer of our common stock shall result in a requirement for us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, (1) the expression an “offer of common stock to the public” in relation to any common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase or subscribe for the common stock, as the same may be varied in that Relevant Member State by any measure implementing the Prospectus Directive in that Relevant Member State, (2) the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive), and includes any relevant implementing measure in each Relevant Member State and (3) the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
This prospectus has only been communicated or caused to have been communicated and will only be communicated or caused to be communicated as an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act of 2000 (the “FSMA”)) as received in connection with the issue or sale of the common stock in circumstances in which Section 21(1) of the FSMA does not apply to us. All applicable provisions of the FSMA will be complied with in respect to anything done in relation to the common stock in, from or otherwise involving the United Kingdom.
Canada
The securities may be sold only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
124
Australia
No placement document, prospectus, product disclosure statement or other disclosure document has been lodged with the Australian Securities and Investments Commission in relation to the offering. This prospectus does not constitute a prospectus, product disclosure statement or other disclosure document under the Corporations Act 2001, or the Corporations Act, and does not purport to include the information required for a prospectus, product disclosure statement or other disclosure document under the Corporations Act.
Any offer in Australia of the securities may only be made to persons, or the Exempt Investors, who are “sophisticated investors” (within the meaning of section 708(8) of the Corporations Act), “professional investors” (within the meaning of section 708(11) of the Corporations Act) or otherwise pursuant to one or more exemptions contained in section 708 of the Corporations Act so that it is lawful to offer the securities without disclosure to investors under Chapter 6D of the Corporations Act.
The securities applied for by Exempt Investors in Australia must not be offered for sale in Australia in the period of 12 months after the date of allotment under the offering, except in circumstances where disclosure to investors under Chapter 6D of the Corporations Act would not be required pursuant to an exemption under section 708 of the Corporations Act or otherwise or where the offer is pursuant to a disclosure document which complies with Chapter 6D of the Corporations Act. Any person acquiring securities must observe such Australian on-sale restrictions.
This prospectus contains general information only and does not take account of the investment objectives, financial situation or needs of any particular person. It does not contain any securities recommendations or financial product advice. Before making an investment decision, investors need to consider whether the information in this prospectus is appropriate to their needs, objectives and circumstances and, if necessary, seek expert advice on those matters.
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange, or SIX, or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the securities or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the securities have been or will be filed with or approved by any Swiss regulatory authority. This document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of securities has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes, or CISA. The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of securities.
Dubai International Financial Centre
This prospectus relates to an Exempt Offer in accordance with the Offered Securities Rules of the Dubai Financial Services Authority, or DFSA. This prospectus is intended for distribution only to persons of a type specified in the Offered Securities Rules of the DFSA. It must not be delivered to, or relied on by, any other person. The DFSA has no responsibility for reviewing or verifying any documents relating to Exempt Offers. The DFSA has not approved this prospectus nor taken steps to verify the information set forth herein and has no responsibility for the prospectus. The securities to which this prospectus relates may be illiquid and/or subject to restrictions on their resale. Prospective purchasers of the securities offered should conduct their own due diligence on the securities. If you do not understand the contents of this prospectus, you should consult an authorized financial advisor.
125
Hong Kong
The securities have not been offered or sold and will not be offered or sold in Hong Kong, by means of any document, other than (a) to “professional investors” as defined in the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance or (b) in other circumstances which do not result in the document being a “prospectus” as defined in the Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance. No advertisement, invitation or document relating to the securities has been or may be issued or has been or may be in the possession of any person for the purposes of issue, whether in Hong Kong or elsewhere, which is directed at, or the contents of which are likely to be accessed or read by, the public of Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to securities which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” as defined in the Securities and Futures Ordinance and any rules made under that Ordinance.
Japan
The securities have not been and will not be registered under the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948, as amended) and, accordingly, will not be offered or sold, directly or indirectly, in Japan, or for the benefit of any Japanese Person or to others for re-offering or resale, directly or indirectly, in Japan or to any Japanese Person, except in compliance with all applicable laws, regulations and ministerial guidelines promulgated by relevant Japanese governmental or regulatory authorities in effect at the relevant time. For the purposes of this paragraph, “Japanese Person” shall mean any person resident in Japan, including any corporation or other entity organized under the laws of Japan.
126
The validity of the issuance of the shares of common stock to be sold in this offering will be passed upon for us by McDermott Will & Emery LLP, New York, New York. Certain legal matters relating to this offering will be passed upon for the underwriters by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., New York, New York.
The balance sheets of Intensity Therapeutics, Inc. as of December 31, 2019 and 2020, and the related statements of operations, changes in redeemable convertible preferred stock and stockholders’ deficiency and cash flows for the years ended December 31, 2019 and 2020, appearing in this prospectus and registration statement, have been audited by EisnerAmper LLP, independent registered public accounting firm, as set forth in their report thereon appearing elsewhere herein, and such report includes explanatory paragraphs on (1) existence of substantial doubt about the Company’s ability to continue as a going concern, and (2) the adoption of Accounting Standards Update 2016-02 “Leases”, and are included herein in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock offered by this prospectus. This prospectus, which constitutes a part of the registration statement, does not contain all the information set forth in the registration statement, some of which is contained in exhibits to the registration statement as permitted by the rules and regulations of the SEC. For further information with respect to us and our common stock, we refer you to the registration statement, including the exhibits filed as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other document are not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, please see the copy of the contract or document that has been filed. Each statement in this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit. The SEC maintains an internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
Upon the completion of this offering, we will be subject to the information reporting requirements of the Exchange Act, and we will file reports, proxy statements and other information with the SEC. These reports, proxy statements and other information will be available at www.sec.gov.
We also maintain a website at www.intensitytherapeutics.com. Information contained in, or accessible through, our website is not a part of this prospectus, and the inclusion of our website address in this prospectus is only as an inactive textual reference.
127
INTENSITY THERAPEUTICS, INC.
INDEX TO FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Intensity Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Intensity Therapeutics, Inc. (the “Company”) as of December 31, 2020 and 2019, and the related statements of operations, changes in redeemable convertible preferred stock and stockholders’ deficiency, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note B to the financial statements, the Company has incurred losses from operations and negative cash flows that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note B. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Change in Accounting Principle
As discussed in Notes C[11] and J to the financial statements, the Company has changed its method of accounting for leases in 2019 due to the adoption of Accounting Standards Update 2016-02 “Leases” (Topic 842).
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2017
EISNERAMPER LLP
New York, New York
September 20, 2021
F-2
INTENSITY THERAPEUTICS, INC.
Balance Sheets
|
December 31, |
||||||||
|
2020 |
2019 |
|||||||
|
ASSETS |
|
|
|
|
||||
|
Current assets: |
|
|
|
|
||||
|
Cash and cash equivalents |
$ |
9,316,092 |
|
$ |
3,828,838 |
|
||
|
Investments |
|
— |
|
|
4,564,813 |
|
||
|
Other current assets |
|
311,870 |
|
|
255,574 |
|
||
|
Total current assets |
|
9,627,962 |
|
|
8,649,225 |
|
||
|
Right-of-use asset, net |
|
490,240 |
|
|
252,668 |
|
||
|
Other assets |
|
31,703 |
|
|
29,441 |
|
||
|
Total assets |
$ |
10,149,905 |
|
$ |
8,931,334 |
|
||
|
|
|
|
|
|||||
|
LIABILITIES, REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIENCY |
|
|
|
|
||||
|
Current liabilities: |
|
|
|
|
||||
|
Accounts payable |
$ |
222,707 |
|
$ |
393,963 |
|
||
|
Accrued expenses |
|
952,043 |
|
|
617,068 |
|
||
|
Current lease liability |
|
171,226 |
|
|
117,194 |
|
||
|
Total current liabilities |
|
1,345,976 |
|
|
1,128,225 |
|
||
|
Long-term liabilities: |
|
|
|
|
||||
|
Related party deposit |
|
36,000 |
|
|
36,000 |
|
||
|
Long-term lease liability |
|
326,255 |
|
|
143,076 |
|
||
|
Total long-term liabilities |
|
362,255 |
|
|
179,076 |
|
||
|
Total liabilities |
|
1,708,231 |
|
|
1,307,301 |
|
||
|
Series A redeemable convertible preferred stock, par value $.0001. Authorized, issued, and outstanding shares of 5,000,000 as of December 31, 2020 and 2019. Liquidation preference of $20,000,000 |
|
10,000,000 |
|
|
10,000,000 |
|
||
|
STOCKHOLDERS’ DEFICIENCY |
|
|
|
|
||||
|
Series B convertible preferred stocks, par value $.0001. Authorized, issued, and outstanding shares of 1,449,113 as of December 31, 2020 and 2019. Liquidation preference of $3,260,504 as of December 31, 2020. |
|
145 |
|
|
145 |
|
||
|
Series C convertible preferred stocks, par value $.0001. Authorized shares of 1,800,606 and 6,000,000 at December 31, 2020 and 2019, respectively. Issued and outstanding shares of 1,800,606 and 695,653 at December 31, 2020 and 2019, respectively. Liquidation preference of $4,051,364 as of December 31, 2020. |
|
180 |
|
|
70 |
|
||
|
Common stock, par value $.0001. Authorized shares of 50,000,000 and 30,000,000 at December 31, 2020 and 2019, respectively. Issued and outstanding shares of 6,820,211 and 6,805,994 at December 31, 2020 and 2019, respectively. |
|
682 |
|
|
681 |
|
||
|
Additional paid in capital |
|
21,666,178 |
|
|
14,843,168 |
|
||
|
Note receivable for purchase of common stock: related party |
|
(50,000 |
) |
|
(75,000 |
) |
||
|
Accumulated deficit |
|
(23,175,511 |
) |
|
(17,145,031 |
) |
||
|
Total stockholders’ deficiency |
|
(1,558,326 |
) |
|
(2,375,967 |
) |
||
|
Total liabilities, redeemable convertible preferred stock and stockholders’ deficiency |
$ |
10,149,905 |
|
$ |
8,931,334 |
|
||
The accompanying notes are an integral part of these financial statements
F-3
INTENSITY THERAPEUTICS, INC.
Statements of Operations
|
Year Ended |
||||||||
|
2020 |
2019 |
|||||||
|
Operating expenses: |
|
|
|
|
||||
|
Research and development costs |
$ |
5,050,565 |
|
$ |
4,437,242 |
|
||
|
General and administrative costs |
|
1,172,681 |
|
|
1,238,290 |
|
||
|
Total operating expenses |
|
6,223,246 |
|
|
5,675,532 |
|
||
|
Loss from operations |
|
(6,223,246 |
) |
|
(5,675,532 |
) |
||
|
Other income: |
|
|
|
|
||||
|
Interest income |
|
74,009 |
|
|
150,707 |
|
||
|
Other |
|
118,757 |
|
|
144,989 |
|
||
|
Net loss |
$ |
(6,030,480 |
) |
$ |
(5,379,836 |
) |
||
|
Loss per share |
|
|
|
|
||||
|
Loss per share, basic and diluted |
$ |
(0.88 |
) |
$ |
(0.79 |
) |
||
|
Weighted average number of shares of common stock, basic and diluted. |
|
6,819,026 |
|
|
6,805,994 |
|
||
The accompanying notes are an integral part of these financial statements
F-4
INTENSITY THERAPEUTICS, INC.
Statements of Changes in Redeemable Convertible Preferred Stock and Stockholders’ Deficiency
Years Ended December 31, 2020 and 2019
|
Series A Redeemable |
Series B |
Series C |
Common Stock |
Additional |
Note |
Accumulated |
Stockholders’ Deficiency |
|||||||||||||||||||||||||||
|
Shares |
Amount |
Shares |
Amount |
Shares |
Amount |
Shares |
Amount |
|||||||||||||||||||||||||||
|
Balances at December 31, 2018 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
— |
$ |
— |
6,805,994 |
$ |
681 |
$ |
10,423,201 |
$ |
(75,000 |
) |
$ |
(11,758,649 |
) |
$ |
(1,409,622) |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
|
Cumulative effect adjustments to accrue deficit related to the adoption of ASU 2016-02 “Leases” |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(6,546) |
|
|
(6,546) |
||||||||||||
|
Balances at January 1, 2019 |
5,000,000 |
|
10,000,000 |
1,449,113 |
|
145 |
— |
|
— |
6,805,994 |
|
681 |
|
10,423,201 |
|
(75,000 |
) |
|
(11,765,195) |
|
|
(1,416,168) |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
|
Stock-based compensation expense |
|
|
|
|
|
561,377 |
|
|
|
|
|
561,377 |
||||||||||||||||||||||
|
Issuance of Series C convertible preferred stock and preferred stock, and/or common stock warrants – net of placement fees of $141,345 |
|
|
695,653 |
|
70 |
|
|
3,858,590 |
|
|
|
|
|
3,858,660 |
||||||||||||||||||||
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(5,379,836) |
|
|
(5,379,836) |
||||||||||||
|
Balances at December 31, 2019 |
5,000,000 |
|
10,000,000 |
1,449,113 |
|
145 |
695,653 |
|
70 |
6,805,994 |
|
681 |
|
14,843,168 |
|
(75,000 |
) |
|
(17,145,031) |
|
|
(2,375,967) |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
|
Stock-based compensation expense |
|
|
|
|
|
555,488 |
|
|
|
|
|
555,488 |
||||||||||||||||||||||
|
Issuance of Series C convertible preferred stock – net of placement fees of $167,591 |
|
|
1,104,953 |
|
110 |
14,217 |
|
1 |
|
6,267,522 |
|
|
|
|
|
6,267,633 |
||||||||||||||||||
|
Repayment on shareholder note |
|
|
|
|
|
|
25,000 |
|
|
|
|
25,000 |
||||||||||||||||||||||
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(6,030,480) |
|
|
(6,030,480) |
||||||||||||
|
Balances at December 31, 2020 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
1,800,606 |
$ |
180 |
6,820,211 |
$ |
682 |
$ |
21,666,178 |
$ |
(50,000 |
) |
$ |
(23,175,511) |
|
$ |
(1,558,326) |
||||||||||||
The accompanying notes are an integral part of these financial statements
F-5
INTENSITY THERAPEUTICS, INC.
Statements of Cash Flows
|
Year Ended |
||||||||
|
2020 |
2019 |
|||||||
|
Cash flows from operating activities: |
|
|
|
|
||||
|
Net loss |
$ |
(6,030,480 |
) |
$ |
(5,379,836 |
) |
||
|
|
|
|
|
|||||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
||||
|
Reduction in carrying amount of right-of-use asset |
|
116,833 |
|
|
95,206 |
|
||
|
Stock-based compensation |
|
555,488 |
|
|
561,377 |
|
||
|
Subtotal of non-cash expenses |
|
672,321 |
|
|
656,583 |
|
||
|
Changes in operating assets and liabilities, net: |
|
|
|
|
||||
|
Other current assets |
|
(56,296 |
) |
|
(27,646 |
) |
||
|
Other assets |
|
(2,262 |
) |
|
(2,526 |
) |
||
|
Accounts payable |
|
(171,256 |
) |
|
202,552 |
|
||
|
Accrued expenses |
|
334,975 |
|
|
228,678 |
|
||
|
Change in lease liabilities |
|
(117,194 |
) |
|
(94,150 |
) |
||
|
Subtotal of changes in operating assets and liabilities |
|
(12,033 |
) |
|
306,908 |
|
||
|
Net cash used in operating activities |
|
(5,370,192 |
) |
|
(4,416,345 |
) |
||
|
|
|
|
|
|||||
|
Cash flows from investing activities: |
|
|
|
|
||||
|
Purchase of short term investments |
|
(10,897,586 |
) |
|
(7,071,416 |
) |
||
|
Redemptions of short term investments |
|
15,462,399 |
|
|
8,768,183 |
|
||
|
Net cash provided by investing activities |
|
4,564,813 |
|
|
1,696,767 |
|
||
|
|
|
|
|
|||||
|
Cash flows from financing activities: |
|
|
|
|
||||
|
Payment received under shareholder note |
|
25,000 |
|
|
— |
|
||
|
Proceeds from sale of preferred stock and preferred stock and/or common stock warrants, net |
|
6,267,633 |
|
|
3,858,660 |
|
||
|
Net cash provided by financing activities |
|
6,292,633 |
|
|
3,858,660 |
|
||
|
|
|
|
|
|||||
|
Net increase in cash and cash equivalents |
|
5,487,254 |
|
|
1,139,082 |
|
||
|
Cash and cash equivalents at beginning of year |
|
3,828,838 |
|
|
2,689,756 |
|
||
|
Cash and cash equivalents at end of year |
$ |
9,316,092 |
|
$ |
3,828,838 |
|
||
|
|
|
|
|
|||||
|
Supplemental disclosure of non-cash financing activities: |
|
|
|
|
||||
|
Right-of-use assets obtained in exchange for new operating lease |
$ |
354,405 |
|
$ |
347,874 |
|
||
|
|
|
|
|
|||||
|
Common stock issued for services to placement agent. |
$ |
81,748 |
|
|
— |
|
||
The accompanying notes are an integral part of these financial statements
F-6
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note A — Nature of Business
[1] Corporate history:
Intensity Therapeutics, Inc. (“the Company”) is a Connecticut based biotechnology company whose treatment approach addresses both the regional and systemic nature of a patient’s cancer. The Company’s DfuseRxSM technology platform has identified a lead drug, INT230-6.
[2] Propriety products and technology portfolios:
The Company’s Phase 1/2 protocol has been authorized to proceed by both the United States Food & Drug Administration (“FDA”) and Health Canada for INT230-6. In May 2017, the Company began the clinical study.
In April 2019, the FDA granted Fast Track designation to the Company’s development program evaluating INT230-6 for the treatment of patients with relapsed or metastatic triple negative breast cancer who have failed at least two prior lines of therapy.
In June 2019, the Company entered into an agreement with Merck (known as MSD outside the United States and Canada), through a subsidiary of Merck, to evaluate the combination of the Company’s lead product candidate INT230-6 and KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 (programmed death receptor-1) therapy, in patients with advanced solid malignancies including pancreatic, bile duct, squamous cell and non-MSI high colon cancers. The Company dosed its first patient in this combination study in October 2019.
In April 2020, the Company entered into a clinical trial collaboration agreement with Bristol Myers Squibb (NYSE: BMY) Company to evaluate the safety and efficacy of our INT230-6 with BMY’s Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) immune checkpoint inhibitor Yervoy® (ipilimumab). The combination will be evaluated in patients with breast cancer, liver cancer and advanced sarcoma.
The Company is now in Phase 2 of the clinical trial.
Since the KEYTRUDA injections may continue for up to two years from the initial dose, the Company anticipates that Phase 1/2 drug testing could continue into 2024.
In March 2021, the Company began the INVINCIBLE study (IT-02), which is a Phase 2 Randomized, Window of Opportunity Trial in Early Stage Breast Cancer. The Company anticipates that this study will be completed in 2022.
Note B — Liquidity and Plan of Operation
The accompanying financial statements have been prepared in conformity with generally accepted accounting principles, which contemplate continuation of the Company as a going concern.
The Company is a development stage company and has not generated any revenue from its product candidates. The Company, therefore, has experienced net losses and negative cash flows from operations each year since its inception. Through December 31, 2020, the Company has an accumulated deficit of approximately $23.2 million. The Company’s operations have been financed primarily through the sale of equity securities. The Company’s net loss for the years ended December 31, 2020 and 2019 were approximately $6.0 million and $5.4 million, respectively.
To date, the Company has not obtained regulatory approval for any of its product candidates. The Company expects to incur significant expenses to complete development of its product candidates. The Company may never be able to obtain regulatory approval for the marketing of any of its product candidates in the United States or internationally and there can be no assurance that the Company will generate revenues or ever achieve profitability. The Company does not expect to receive significant product revenue in the near term. The Company, therefore, expects to continue to incur substantial losses for the foreseeable future.
Cash and cash equivalents at December 31, 2020 totaled approximately $9.3 million. Until such time, if ever, as the Company can generate substantial product revenue, the Company expects to finance its cash needs through a combination of equity offerings and debt financings. The Company does not have any committed external source
F-7
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note B — Liquidity and Plan of Operation (cont.)
of funds. To the extent that the Company can raise additional capital through the sale of equity or convertible debt securities, the ownership interest of the Company stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of a common stockholder. If the Company is unable to raise additional funds through equity or debt financings when needed, the Company may be required to delay, limit, reduce or terminate its research and product development.
Based on cash on hand at December 31, 2020, the Company believes that it will have sufficient cash to fund planned operations through June 30, 2022. However, the acceleration or reduction of cash outflows by management can significantly impact the timing for raising additional capital to complete development of its product candidates. To continue development, the Company will need to raise additional capital through debt and/or equity financing. Additional capital may not be available on terms favorable to the Company, if at all. The Company does not know if its future offerings will succeed. Accordingly, no assurances can be given that management will be successful in these endeavors. The Company’s recurring losses from operations have caused management to determine there is substantial doubt about its ability to continue as a going concern. These financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities or any other adjustments that might be necessary should the Company be unable to continue as a going concern.
Note C — Summary of Significant Accounting Policies and Accounts
[1] Basis of presentation:
The accompanying financial statements include the accounts of Intensity Therapeutics, Inc. The Company neither owns nor controls any subsidiary companies. The accompanying financial statements have been prepared by the Company in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and reflect the operations of the Company.
[2] Use of estimates:
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Certain accounting principles require subjective and complex judgments to be used in the preparation of financial statements. Accordingly, a different financial presentation could result depending on the judgments, estimates, or assumptions that are used.
The Company utilizes significant estimates and assumptions in valuing its stock-based compensation awards. An additional significant estimate is that these financial statements are based on the assumption of the Company continuing as a going concern. See note B with regard to the Company’s ability to continue as a going concern.
[3] Correction of Immaterial Errors and Adoption of New Lease Accounting Standard:
During the preparation of these financial statements as of and for the year ended December 31, 2020, management corrected its previously issued financial statements for immaterial accounting errors. Specifically, the Company previously expensed legal fees associated with the issuance of equity instead of recording these costs directly to Additional Paid in Capital in the Equity section of the Balance Sheet. The Company previously used incorrect variables in the calculation of stock based compensation. Additionally, the Company adopted Accounting Standards Update 2016-02 “Leases” (Topic 842) (“ASU 2016-12 “Leases”).
F-8
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
Accordingly, the following tables summarize the effects of the immaterial error corrections and the adaption of ASU 2016-02 “Leases” to the Company’s financial statements and the adoption of the new lease accounting standard on January 1, 2019.
At December 31, 2019:
|
As |
Impact of |
Impact of |
Impact of |
Impact of |
Impact of |
As |
||||||||||||||||||||||
|
Balance Sheet |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
Right-of-use asset, net |
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
252,668 |
|
$ |
252,668 |
|
|||||||
|
Total assets |
|
8,678,666 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
252,668 |
|
|
8,931,334 |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
Current lease liability |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
101,951 |
|
|
101,951 |
|
|||||||
|
Total current liabilities |
|
1,011,031 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
101,951 |
|
|
1,112,982 |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
Long-term lease liability |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
158,319 |
|
|
158,319 |
|
|||||||
|
Total long-term liabilities |
|
36,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
158,319 |
|
|
194,319 |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
Total liabilities |
|
1,047,031 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
260,270 |
|
|
1,307,301 |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
Additional paid in capital |
|
15,167,632 |
|
|
(61,783 |
) |
|
(119,056 |
) |
|
(61,345 |
) |
|
(82,280 |
) |
|
— |
|
|
14,843,168 |
|
|||||||
|
Accumulated deficit |
|
(17,461,893 |
) |
|
61,783 |
|
|
119,056 |
|
|
61,345 |
|
|
82,280 |
|
|
(7,602 |
) |
|
(17,145,031 |
) |
|||||||
|
Total Stockholders’ deficiency |
|
($2,368,365 |
) |
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
(7,602 |
) |
$ |
(2,375,967 |
) |
|||||||
At December 31, 2020:
|
As |
Impact of |
Impact of |
Impact of |
Impact of |
Impact of |
Impact of |
As |
||||||||||||||||||||||||
|
Balance Sheet |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
Right-of-use asset, net |
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
252,668 |
|
$ |
237,572 |
$ |
490,240 |
|
||||||||
|
Total assets |
|
9,659,665 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
252,668 |
|
|
237,572 |
|
10,149,905 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
Current lease liability |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
101,951 |
|
|
69,275 |
|
171,226 |
|
||||||||
|
Total current liabilities |
|
1,174,750 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
101,951 |
|
|
69,275 |
|
1,345,976 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
Long-term lease liability |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
158,319 |
|
|
167,936 |
|
326,255 |
|
||||||||
|
Total long-term liabilities |
|
36,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
158,319 |
|
|
167,936 |
|
362,255 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
Total liabilities |
|
1,210,750 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
260,270 |
|
|
237,211 |
|
1,708,231 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
Additional paid in capital |
|
21,948,536 |
|
|
(123,128 |
) |
|
(201,336 |
) |
|
(35,844 |
) |
|
77,950 |
|
|
— |
|
|
— |
|
21,666,178 |
|
||||||||
|
Accumulated deficit |
|
(23,450,628 |
) |
|
123,128 |
|
|
201,336 |
|
|
35,844 |
|
|
(77,950 |
) |
|
(7,602 |
) |
|
361 |
|
(23,175,511 |
) |
||||||||
|
Total Stockholders’ deficiency |
|
($1,551,085 |
) |
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
(7,602 |
) |
$ |
361 |
$ |
(1,558,326 |
) |
||||||||
F-9
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
Year ended December 31, 2019:
|
As |
Impact of |
Impact of |
Impact of |
As |
|||||||||||||
|
Statement of Operations |
|
|
|
|
|
|
|
||||||||||
|
Total Research and development costs |
$ |
4,519,522 |
|
— |
|
$ |
(82,280 |
) |
|
— |
$ |
4,437,242 |
|||||
|
|
|
|
|
|
|
|
|||||||||||
|
Total General and administrative costs |
|
1,298,579 |
|
(61,345 |
) |
|
— |
|
|
1,056 |
|
1,238,290 |
|||||
|
|
|
|
|
|
|
|
|||||||||||
|
Net loss |
$ |
5,522,405 |
$ |
(61,345 |
) |
$ |
(82,280 |
) |
$ |
1,056 |
$ |
5,379,836 |
|||||
Year ended December 31, 2020:
|
As |
Impact of |
Impact of |
Impact of |
As |
|||||||||||||
|
Statement of Operations |
|
|
|
|
|
|
|
||||||||||
|
Total Research and development costs |
$ |
4,972,615 |
|
— |
|
$ |
77,950 |
|
— |
|
$ |
5,050,565 |
|||||
|
|
|
|
|
|
|
|
|||||||||||
|
Total General and administrative costs |
|
1,208,886 |
|
(35,844 |
) |
|
— |
|
(361 |
) |
|
1,172,681 |
|||||
|
|
|
|
|
|
|
|
|||||||||||
|
Net loss |
$ |
5,988,735 |
$ |
(35,844 |
) |
$ |
77,950 |
$ |
(361 |
) |
$ |
6,030,480 |
|||||
[4] Concentration of credit risk:
The Company’s financial instruments that are exposed to concentrations of credit risk consist entirely of cash, certificates of deposit, and investments in U.S. Treasury bills. These financial instruments are held at two major U.S. financial institutions. The cash accounts and certificates of deposit are insured by the Federal Deposit Insurance Corporation (“FDIC”) up to regulatory limits. At all times throughout the years ended December 31, 2020 and 2019, the Company’s cash and certificate of deposit balances exceeded the FDIC insurance limit. The Company has not experienced any losses in such accounts. The investments in U.S. Treasury bills are not FDIC insured, but are backed by the U.S. government. U.S. Treasury bills are subject to market risk if they are sold prior to maturity.
[5] Cash and cash equivalents:
The Company considers all liquid investments with an original maturity of three months or less to be cash equivalents.
F-10
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
[6] Fair value measurement:
The Company reports its investments at fair value. Fair value is an estimate of the exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants (i.e., the exit price at the measurement date). Fair value measurements are not adjusted for transaction costs. A fair value hierarchy provides for prioritizing inputs to valuation techniques used to measure fair value into three levels:
|
Level 1 |
Unadjusted quoted prices in active markets for identical assets or liabilities. |
|
|
Level 2 |
Inputs other than quoted market prices that are observable, either directly or indirectly, and reasonably available. Observable inputs reflect the assumptions market participants would use in pricing the asset or liability and are developed based on market data obtained from sources independent of the Company. |
|
|
Level 3 |
Unobservable inputs. Unobservable inputs reflect the assumptions that the Company develops based on available information about what market participants would use in valuing the asset or liability. |
An asset’s or liability’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Availability of observable inputs can vary and is affected by a variety of factors. The Company uses judgment in determining fair value of assets and liabilities and Level 3 assets and liabilities involve greater judgment than Level 1 or Level 2 assets or liabilities.
At December 31, 2019, the Company has investments of $4,564,813 in U.S. Treasury Bills. U.S. government securities are valued using a model that incorporates market observable data, such as reported sales of similar securities, broker quotes, yields, bids, offers, and reference data. U.S. Treasury Bills are categorized in Level 1 of the fair value hierarchy. At December 31, 2020 there were no investments in US Treasury Bills.
The Company’s financial instruments, including cash equivalents, investments and current liabilities are carried at cost, which approximates fair value due to the short-term nature of these instruments.
[7] Stock-based compensation:
Effective January 1, 2020, upon the adoptions of ASU 2018-07, Compensation — Stock Compensation: Improvements to Nonemployee Share-Based Payment Accounting, the Company accounts for stock-based compensation to employees and non-employees in conformity with the provisions of Accounting Standards Codification (“ASC”) ASC Topic 718, “Compensation — Stock Compensation”. Stock compensation consists of stock option grants that were recognized in the statements of operations based on their fair values at the date of grant.
Prior to January 1, 2020, the Company accounted for equity instruments issued to non-employees effective January 1, 2020 in accordance with the provisions of ASC Topic 505, “Equity-Based Payments to Non-Employees” based upon the fair value of the underlying instrument. The equity instruments consisted primarily of stock options. The fair value of such awards was subject to periodic adjustment as the underlying equity instruments vest and was recognized as expense over the period services were received.
The Company calculates the fair value of option grants utilizing the Black-Scholes pricing model. The amount of stock-based compensation recognized during a period is based on the value of the portion of the awards that are ultimately expected to vest. The authoritative guidance requires forfeitures to be estimated at the time stock options are granted and warrants are issued and revised. If necessary in subsequent periods, an adjustment will be booked if actual forfeitures differ from those estimated. The term “forfeitures” is distinct from “cancellations” or “expirations” and represents only the unvested portion of the surrendered stock option or warrant. The Company estimates forfeiture rates for all unvested awards when calculating the expense for the period. In estimating the forfeiture rate, the Company monitors both stock option and warrant exercises as well as employee and non-employee termination patterns.
The resulting stock-based compensation expense for both employee and non-employee awards is generally recognized on a straight-line basis over the requisite service period of the award.
F-11
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
[8] Research and development and patent costs:
Research and development costs are charged to operations as they are incurred. Legal fees and other direct costs incurred in obtaining and protecting patents are also expensed as incurred, due to the uncertainty with respect to future cash flows resulting from the patents and are included as part of general and administrative expenses in the Company’s Statements of Operations.
[9] Income taxes:
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. ASC 740 prescribes the use of the asset-and-liability method whereby deferred tax assets and liabilities are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company utilizes a valuation allowance to reduce deferred tax assets to their estimated realizable value.
The Company accounts for uncertain tax positions in accordance with the provisions of ASC 740. When uncertain tax positions exist, the Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely than not be realized.
The determination as to whether the tax benefit will more likely than not be realized is based upon the technical merits of the tax position as well as consideration of the available facts and circumstances. At
December 31, 2020 and 2019, the Company does not have any significant uncertain tax positions.
There are no estimated interest costs and penalties provided for in the Company’s financial statements for the years ended December 31, 2020 and 2019. If at any time the Company should record interest and penalties in connection with an uncertain tax position, the interest and penalties will be expensed within the income tax line.
The Company’s income tax returns are subject to Federal, state and local income tax examination by the authorities for the last three tax years.
[10] Stock issuance costs:
For the years ended December 31, 2020 and 2019, the Company had incurred approximately $167,600 and $141,300 of costs related to the sale of the Series C preferred stock. These costs were recorded as a deduction to Additional Paid in Capital.
[11] Leases:
The Company determines if an arrangement contains a lease at contract inception. With the exception of short-term leases (leases with terms less than 12 months), all leases with contractual fixed costs are recorded on the balance sheet on the commencement date as a right-of-use (ROU) asset and a lease liability. Lease liabilities to be paid over the next twelve months are classified as current lease liability and all other lease obligations are classified as Long-term lease liability. Lease liabilities are initially measured at the present value of the future minimum lease payments and subsequently increased to reflect the interest accrued and reduced by the lease payments made. The Company’s building leases require a pro-rata share of operating expense and real estate taxes, which are variable in nature and excluded from the measurement of lease liabilities. ROU assets are initially measured at the present value of the future minimum lease payments adjusted for any prior lease pre-payments, lease incentives and initial direct costs. Certain leases contain escalation, renewal and/or termination options that are factored into the ROU asset as appropriate. Operating leases result in a straight-line rent expense over the expected lease term.
F-12
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
The Company uses its estimated incremental borrowing rate, which is derived from information available at the lease commencement date, in determining the present value of future lease payments, if the rate implicit in the lease is not readily determinable. Consideration is given to publicly available data for instruments with similar characteristics when calculating incremental borrowing rates. This incremental borrowing rate estimate is based on a synthetic credit rating derived from the market capitalization of similar companies, the treasury yield curve, and corporate yield spreads.
[12] Basic and dilutive loss per share:
Basic net loss per share is determined using the weighted average number of shares of common stock outstanding during each period. Dilutive net loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as convertible preferred stock, stock options, and stock warrants, which would result in the issuance of incremental shares of common stock. The computation of diluted net loss per share does not include the conversion of securities that would have an anti-dilutive effect. Potential common shares issuable upon conversion of preferred stock, exercise of stock options, and exercise of warrants that are excluded from the computation of diluted weighted average shares outstanding are 9,723,969 and 8,394,642 as of December 31, 2020 and 2019, respectively. The basic and diluted computation of net loss per share for the Company are the same because the effects of the Company’s convertible securities would be anti-dilutive. All common and preferred stock participate equally in dividends and the distribution of earnings if and when declared by the Board of Directors, on the Company’s common stock. For purposes of computing earnings per share, all series of preferred stock are considered participating securities. Therefore, the Company must calculate basic and diluted earnings per share using the two-class method. Under the two-class method, net income for the period is allocated between common stockholders and participating securities according to dividends declared and participation rights in undistributed earnings. As the preferred shareholders have no obligation to fund losses no portion of net loss was allocated to the participating securities for the years ended December 31, 2020 and 2019.
At December 31, 2020 and 2019, the following are shares excluded from the computation of diluted weighted average shares outstanding:
|
December 31, 2020 |
December 31, 2019 |
|||
|
Preferred stock Series A issued |
5,000,000 |
5,000,000 |
||
|
Preferred stock Series B issued |
1,449,113 |
1,449,113 |
||
|
Preferred stock Series C issued |
1,800,606 |
695,653 |
||
|
Options outstanding |
1,394,500 |
1,347,000 |
||
|
Warrants outstanding |
586,500 |
243,000 |
||
|
10,230,719 |
8,734,766 |
[13] Recent accounting pronouncements:
In February 2016, the Financial Accounting Standards Board issued ASU 2016-02 “Leases” which requires lessees to recognize the assets and liabilities for leases with lease terms of more than 12 months and disclose key information about leasing arrangements. The guidance for lessors is largely unchanged from current U.S. GAAP.
The Company adopted ASU 2016-02 “Leases” effective January 1, 2019 using the modified retrospective approach. This approach allows the Company to initially apply the accounting standards at the adoption date and recognize a cumulative adjustment to the opening balance of retained earnings in the period of adoption. The cumulative adjustment at January 1, 2019 was $6,546. The Company elected the package of practical expedients permitted under the transition guidance within the new standard, which among other things, allowed the Company to carry forward the historical lease classification.
Adoption of ASU 2016-02 “Leases” required the Company to restate amounts as of January 1, 2019, resulting in an increase in operating lease right-of-use assets of $347,874 and an increase in lease liabilities of $354,420.
F-13
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
In June 2018, the FASB issued ASU No. 2018-07, “Compensation — Stock Compensation: Improvements to Nonemployee Shared-Based Payment Accounting (“Topic 718”)”, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718 to include share-based payments transactions for acquiring goods and services from nonemployees. Some of the areas for simplification apply only to nonpublic entities.
The amendments specify that Topic 718 applies to all share-based payment transactions in which a grantor acquires goods or services to be used or consumed in a grantor’s own operations by issuing share-based payment awards. The amendments also clarify that Topic 718 does not apply to share-based payments used to effectively provide (1) financing to the issuer or (2) awards granted in conjunction with selling goods or services to customers as part of a contract accounted for under Topic 606, “Revenue from Contracts with Customers.” The amendments in this update are effective for nonpublic companies for fiscal years beginning after December 15, 2019. Early adoption is permitted, but no earlier than a Company’s adoption date of Topic 606. The Company adopted this pronouncement as of January 1, 2020, noting no material impact on the Company’s financial statements.
Note D — Other Current Assets
Other current assets at December 31 include:
|
2020 |
2019 |
|||||
|
Advance to subcontractor |
$ |
150,000 |
$ |
150,000 |
||
|
Prepaid insurance |
|
67,174 |
|
55,468 |
||
|
Taxes receivable |
|
53,812 |
|
21,934 |
||
|
Other current assets |
|
40,884 |
|
28,172 |
||
|
$ |
311,870 |
$ |
255,574 |
|||
The $150,000 advance is to a company which provides services in managing the Company’s clinical trials. The advance is intended to offset the normal delay in the Company reimbursing them for their services and for their payments to the Company’s clinical sites.
Note E — Note Receivable
On February 4, 2015, the Company received a $75,000 note from its Chief Medical Officer. The note was used to fund 75% of a common stock purchase. Interest accrues at the rate of three percent per annum, compounding annually on each anniversary of the date of the note creation.
In 2019, the payment terms were amended to the following schedule, unless paid earlier:
• $25,000 due on or before July 11, 2020;
• $25,000 due on or before July 11, 2021; and
• the remaining balance, including the interest due, in full on or before February 4, 2022.
The balance due on this note is $50,000 and $75,000 at December 31, 2020 and 2019, respectively. In July 2021, the Company made a payment of $25,000. In August 2021, the Board approved a bonus to the Chief Medical Officer for $25,000, which was the remaining balance at that time, and the $15,073 of related accrued interest, in order to extinguish the remaining balance outstanding of his note.
F-14
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note E — Note Receivable (cont.)
Sale of the related securities would require a repayment of the note. The note is recorded as a reduction to stockholders’ equity since the note was used to purchase the Company’s common stock. Accrued interest on this note is approximately $14,000 and $11,700 at December 31, 2020 and 2019, respectively, and is included in other assets and interest income.
Note F — Accrued Expenses
Accrued expenses at December 31 include:
|
2020 |
2019 |
|||||
|
Accrued vacation, wages, and related payroll taxes |
$ |
236,150 |
$ |
163,602 |
||
|
Patient costs incurred but not yet invoiced |
|
706,027 |
|
446,950 |
||
|
Accrued other |
|
9,866 |
|
6,516 |
||
|
$ |
952,043 |
$ |
617,068 |
|||
Note G — Stockholders’ Equity
[1] Authorized and outstanding shares:
The total number of shares of all classes of stock which the Company shall have authority to issue, at December 31, in which all shares have a par value of $0.0001 are as follows:
|
2020 |
2019 |
|||
|
Common stock |
50,000,000 |
30,000,000 |
||
|
Preferred stock: |
||||
|
Series A |
5,000,000 |
5,000,000 |
||
|
Series B |
1,449,113 |
1,449,113 |
||
|
Series C |
1,800,606 |
6,000,000 |
||
|
undesignated |
11,750,281 |
5,050,887 |
||
|
Total Preferred stock |
20,000,000 |
17,500,000 |
The Company has issued the following number of shares, options, and warrants:
|
2020 |
2019 |
|||
|
Common shares issued |
6,820,211 |
6,805,994 |
||
|
Preferred stock Series A issued |
5,000,000 |
5,000,000 |
||
|
Preferred stock Series B issued |
1,449,113 |
1,449,113 |
||
|
Preferred stock Series C issued |
1,800,606 |
695,653 |
||
|
Total outstanding shares |
15,069,930 |
13,950,760 |
||
|
Options and warrants outstanding |
1,981,000 |
1,590,000 |
||
|
Total outstanding shares, options and warrants |
17,050,930 |
15,540,760 |
F-15
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note G — Stockholders’ Equity (cont.)
In 2020, the Company amended its 2013 Stock Option Plan to increase the number of authorized shares available under the plan from 1,800,000 to 4,500,000. At December 31, 2020, there are 3,105,500 unissued options reserved for issuance under the Company’s 2013 Stock Option Plan.
In 2019, the Company initiated an offering of Series C Preferred Stock (“Series C”) with a par value of $0.0001, in which the Company was authorized to sell up to 6 million shares. The Company sold 1,800,606 shares (1,104,953 in 2020 and 695,653 in 2019) for net proceeds of $ 10,044,545 ($6,185,885 in 2020 and $3,858,660 in 2019.) In conjunction with the Series C raise, the Company issued warrants which the holder can convert into 60,000 shares of Series C preferred stock or common stock at the discretion of the Company. For purposes of determining value, management used the value of common shares in its calculation. Each share of the preferred stock or common stock under the warrant is converted for $6.25 per share of preferred or common stock. The warrants expire on December 31, 2022. Subsequent to this sale, the number of authorized shares of Series C was reduced to 1,800,606. These warrants are not included in the computation of weighted average number of common stock, basic and diluted. These warrants, if exercised, will participate equally in dividends and distributions of earnings.
Through December 31, 2020, the Company has raised approximately:
|
Sale of common stock (2012 to 2020) |
$ |
3.2 million |
|
|
Sale of Series A preferred stock (2015 to 2016) |
|
10.0 million |
|
|
Sale of Series B preferred stock (2018) |
|
6.5 million |
|
|
Sale of Series C preferred stock (2019 to 2020) |
|
10.4 million |
|
|
Total proceeds from the issuance of stock |
$ |
30.1 million |
[2] Voting:
As long as there are issued and outstanding shares of 2,500,000 or more Preferred Stock, the holders of record of the outstanding shares of Preferred Stock shall be entitled to elect two directors of the Company and the holders of record of the shares of Common Stock shall be entitled to elect three directors of the Company.
There are currently four directors of the Company, and the Board of Directors shall be comprised of no more than five directors.
The holders of the Common Stock are entitled to one vote for each share of Common Stock held at all meetings of stockholders. The holders of the Preferred Stock are entitled to cast the number of votes equal to the number of whole shares of Common Stock into which the shares of Preferred Stock held by such holder are convertible as of the record date at a 1:1 conversation ration, subject to adjustments for stock dividends, splits, combinations, and similar events. Preferred holders vote together with common stockholders as of a single class.
[3] Dividends:
The holders of the Preferred Stock are entitled to the same cash dividend that is paid to holders of Common Stock. The Company has not declared or paid any dividends.
[4] Redemption rights:
At any time on or after May 18, 2022, the holders of at least two-thirds of the then outstanding shares of Series A Preferred Stock may elect to cause the Company to redeem all but not less than all of the shares of Series A Preferred Stock at a redemption price per share in cash equal to $2, the Series A Original Issue Price per Share. Series B and C preferred stockholders do not have any redemption rights.
F-16
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note G — Stockholders’ Equity (cont.)
[5] Liquidation preference:
In the event of any voluntary or involuntary liquidation, the holders of the Series A Preferred Stock shall be entitled to be paid out of the assets of the Corporation before any payment shall be made to the holders of Common Stock, in an amount equal to the greater of (i) two times the Series A Original Issue Price, plus any dividends declared but unpaid thereon, or (ii) such amount per share as would have been payable had all shares of Series A Preferred Stock been converted into Common Stock.
The holders of the Series B and C Preferred Stock shall receive distributions pari passu to holders of Series A stockholders. The holders of Series B and C Preferred Stock shall receive the greater of: (i) in preference to any distribution to holders of Common Stock, $2.25 per share of Series B holders and Series C holders, plus declared and unpaid dividends, if any, on each share of Series B and C Preferred Stock, with the balance of any proceeds then distributed pro rata to holders of Company Stock; or (ii) an amount equal to a pro rata share of the proceeds available for distribution to all holders of Company stock (treating the Series B and C Preferred Stock on an as converted to Common Stock basis).
[6] Conversion:
Each share of Series A, B and C Preferred Stock shall be convertible, at the option of the holder thereof, at any time and from time to time, and without the payment of additional consideration by the holder thereof, into such number of fully paid and non-assessable shares of Common Stock as is determined by dividing the Series Original Issue Price by the Series A, B or C Conversion Price (as defined below) in effect at the time of conversion. The “Series A Conversion Price”, “Series B Conversion Price” and “Series C Conversion Price” shall initially be equal to $2.00, $4.50 and $5.75, respectively. Such initial Conversion Price, and the rate at which shares of Preferred Stock may be converted into shares of Common Stock, shall be subject to adjustments as defined in the Certificate of Incorporation.
Upon either (a) the closing of the sale of shares of Common Stock to the public in a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act of 1933, as amended, resulting in at least $15,000,000 of gross proceeds to the Corporation or (b) the date and time, or the occurrence of an event, specified by vote or written consent of the holders of at least a majority of the then outstanding shares of either Series A, B or C Preferred Stock, then (i) all outstanding shares of either Series A, B or C Preferred Stock shall automatically be converted into shares of Common Stock, at the then effective conversion rate as calculated above, and (ii) such shares may not be reissued by the Corporation.
Note H — Common Stock Warrants
The Company had 586,500 and 243,000 outstanding warrants at December 31, 2020 and 2019, respectively. The weighted average exercise price of warrants outstanding at December 31, 2020 and 2019 was $2.72 and $3.05, respectively. All warrants outstanding at December 31, 2020 and 2019 were exercisable.
F-17
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note H — Common Stock Warrants (cont.)
The following table summarizes information about common stock warrants outstanding and exercisable at December 31, 2020 and 2019:
|
Number |
Weighted |
||||
|
Balance at December 31, 2018 |
183,000 |
$ |
2.00 |
||
|
Issued |
60,000 |
|
6.25 |
||
|
Balance at December 31, 2019 |
243,000 |
|
3.05 |
||
|
Issued |
31,500 |
|
5.75 |
||
|
Options reclassified as warrants |
312,000 |
|
2.16 |
||
|
Balance at December 31, 2020 |
586,500 |
$ |
2.72 |
||
|
Exercisable at December 31, 2020 |
546,668 |
$ |
2.53 |
||
The warrants issued during 2019 relate to the issuance of the Series C Preferred Stock and were valued at approximately $141,000, and vested immediately with an exercise price of $6.25 and expires in 3 years. The warrants issued during 2020 were granted to independent contractors for work that they performed for the Company which were valued at approximately $103,000 which vested immediately with an exercise price of $5.75 and expires in 10 years.
Prior to 2020, the Company had issued 312,000 options to independent contractors. During 2020, these options were changed to warrants with the same terms and conditions as the original options.
The aggregate intrinsic value of outstanding warrants was $1,805,000 and $686,250 at December 31, 2020 and 2019, respectively.
The following table summarizes the assumptions used to estimate the fair value of common stock warrants granted during the years ended December 31, 2020 and 2019:
|
2020 |
2019 |
|||||
|
Stock price |
$ |
5.75 |
$ |
5.75 |
||
|
Exercise price |
$ |
5.75 |
$ |
6.25 |
||
|
Expected volatility |
|
66.60% |
|
63.50% |
||
|
Risk free interest rates |
|
0.55% |
|
1.67% to 3.07% |
||
|
Expected term |
|
10.00 years |
|
3.00 years |
||
For 2020 and 2019, a dividend yield of 0% was used because the Company has not historically paid, and does not intend to pay, a dividend on common stock in the foreseeable future. Based on history, the Company uses a forfeiture rate of 0% based on minimal turnover. The expected stock price volatility was estimated based on the historical volatilities for industry peers, as the Company has no active market for its stock, and limited history for issuance price of its stock.
The risk free rate assumption is determined using the yield currently available on U.S. Treasury zero coupon issues with a remaining term commensurate with the expected term of the award. The expected term of the option represents the period the options are expected to be outstanding.
F-18
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note H — Common Stock Warrants (cont.)
The following table summarizes information about common stock warrants outstanding at December 31, 2020:
|
Exercise Price |
Number |
Weighted |
Number |
Weighted |
||||
|
$1.00 – $1.50 |
120,000 |
3.42 years |
120,000 |
3.42 years |
||||
|
$2.00 – $2.50 |
336,500 |
4.96 years |
336,500 |
4.96 years |
||||
|
$4.00 – $4.50 |
38,500 |
8.00 years |
27,500 |
7.80 years |
||||
|
$5.75 – $6.25 |
91,500 |
4.62 years |
62,668 |
2.32 years |
||||
|
586,500 |
546,668 |
Note I — Stock-Based Compensation
The Company has a stock option plan, the 2013 Stock Option Plan (the “Plan”), which is administered by the Committee. Under the Plan, stock options to purchase a total of 4,500,000 shares of common stock, may be granted to eligible employees, officers, directors, and consultants of the Company.
The Company had outstanding options as follows:
|
Number |
Weighted |
|||||
|
Outstanding at January 1, 2019 |
1,037,000 |
|
$ |
2.47 |
||
|
Issued |
310,000 |
|
$ |
4.50 |
||
|
Outstanding at December 31, 2019 |
1,347,000 |
|
$ |
2.93 |
||
|
Issued |
359,500 |
|
$ |
5.75 |
||
|
Options reclassified as warrants |
(312,000 |
) |
$ |
2.16 |
||
|
Outstanding at December 31, 2020 |
1,394,500 |
|
$ |
3.83 |
||
|
Exercisable December 31, 2020 |
887,750 |
|
$ |
3.03 |
||
The aggregate intrinsic value of options outstanding was $2,410,313 and $3,288,033 at December 31, 2020 and 2019, respectively.
The following table summarizes information about stock options at December 31, 2020:
|
Outstanding Options |
Exercisable Options |
|||||||
|
Exercise Price |
Number |
Weighted |
Number |
Weighted |
||||
|
$1.00 – $1.50 |
200,000 |
3.20 years |
200,000 |
3.20 years |
||||
|
$2.00 |
290,000 |
5.46 years |
282,500 |
5.44 years |
||||
|
$4.00 – $4.50 |
545,000 |
7.97 years |
373,750 |
7.89 years |
||||
|
$5.75 |
359,500 |
9.65 years |
31,500 |
9.61 years |
||||
|
1,394,500 |
887,750 |
|||||||
Employee option vesting is based on the employee’s continued employment with the Company.
F-19
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note I — Stock-Based Compensation (cont.)
In 2018, the Board of Directors amended the 2013 Stock Option Plan to provide an immediate vesting of outstanding options in the event of a change of control, such as an acquisition, notwithstanding any other provision of the Stock Option Plan.
The following table summarizes the assumptions used to estimate the fair value of stock options granted during the years ended December 31, 2020 and 2019:
|
2020 |
2019 |
|||
|
Expected volatility |
67.00% – 69.60% |
63.30% – 63.80% |
||
|
Risk free interest rates |
0.38% to 0.49% |
1.63% to 2.42% |
||
|
Expected term |
4.00 years |
3.00 years |
For 2020 and 2019, a dividend yield of 0% was used because the Company has not historically paid, and does not intend to pay, a dividend on common stock in the foreseeable future. Based on history, the Company uses a forfeiture rate of 0% based on minimal turnover. Forfeitures are recognized as they occur. The expected stock price volatility assumption was estimated based on the historical volatilities for industry peers, as the Company has no active market for its stock, and limited history for issuance price of its stock.
The risk free rate assumption is determined using the yield currently available on U.S. Treasury zero coupon issues with a remaining term commensurate with the expected term of the award. The expected term of the option represents the period the options are expected to be outstanding.
The weighted average grant date fair value of stock options granted was $2.91 and $1.95 for 2020 and 2019, respectively.
All options are granted with an exercise price equal to the current fair market value of the stock. In 2020 and 2019, all options were issued with an exercise price of $5.75 and $4.50, respectively.
At December 31, 2020, total unrecognized compensation cost related to non-vested options was approximately $1,169,000 and is expected to be recognized over the remaining weighted average service period of 3.2 years. The Company recorded stock-based compensation related to stock options of approximately $439,000 and $398,000 within research and development costs for the years ended December 31, 2020 and 2019, respectively. The Company recorded stock-based compensation expense related to stock options of approximately $116,000 and $163,000 within general and administrative costs for the years ended December 31, 2020 and 2019, respectively.
All options expire ten years from date of grant. Options outstanding begin to expire in August 2023. Options that were granted to employees and consultants have vesting periods that vary by award to recipient and range from immediate vesting to a period of up to 4 years.
The shares of stock underlying stock options are restricted securities under U. S. Federal and applicable state securities laws and, as such, may not be transferred, sold, or otherwise disposed of in the United States, except as permitted under U.S. Federal and state securities laws, pursuant to registration or exemption therefrom.
Note J — Leases
In January 2017, the Company entered a lease for 2,534 square feet of office space at its current location. The lease commenced in May 2017. The initial lease term was two years. In November 2018, the Company exercised the option to extend for an additional three years.
F-20
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note J — Leases (cont.)
In July 2020, the Company amended this lease to increase office space by an additional 1,653 square feet in the same building. The amended lease that includes the space included in the original lease has monthly rent as follows:
|
Year 1 (October 2020 through September 2021) |
$15,502.50 per month |
($44.43 per square foot) |
||
|
Year 2 (October 2021 through September 2022) |
$15,851.42 per month |
($45.43 per square foot) |
||
|
Year 3 (October 2022 through September 2023) |
$16,200.33 per month |
($46.43 per square foot) |
The Company has an option to extend this amended lease for an additional 3 years at the following amounts:
|
Year 4: (October 2023 through September 2024) |
$16,338.08 per month |
($46.83 per square foot) |
||
|
Year 5: (October 2024 through September 2025) |
$16,475.83 per month |
($47.22 per square foot) |
||
|
Year 6: (October 2025 through September 2026) |
$16,824.75 per month |
($48.22 per square foot) |
The Company has until October 1, 2023 to exercise this option to extend.
The Company also pays a pro-rata share of operating expenses and real estate taxes.
The following table summarizes the balance sheet classification of the operating lease assets and related lease liabilities at December 31:
|
2020 |
2019 |
|||||
|
Operating lease right-of-use assets |
$ |
490,240 |
$ |
252,668 |
||
|
Current portion of operating lease liabilities |
$ |
171,226 |
$ |
117,194 |
||
|
Long-term operating lease liabilities |
|
326,255 |
|
143,076 |
||
|
$ |
497,481 |
$ |
260,270 |
|||
The following variables were used to determine the right-of-use asset and the operating lease liabilities at December 31:
|
2020 |
2019 |
|||
|
Weighted average remaining operating lease term |
2.75 years |
2.42 years |
||
|
Weighted average operating lease discount rate |
3.92% |
5.38% |
Facilities and rent expense for the year ended December 31 are as follows:
|
2020 |
2019 |
|||||||
|
Operating lease cost |
$ |
130,613 |
|
$ |
111,496 |
|
||
|
Sublease income from related party |
|
(45,444 |
) |
|
(40,136 |
) |
||
|
Net lease cost |
|
85,169 |
|
|
71,360 |
|
||
|
Utilities |
|
8,061 |
|
|
6,628 |
|
||
|
Cleaning, repairs, and other |
|
10,907 |
|
|
19,915 |
|
||
|
Facilities and rent expense |
$ |
104,137 |
|
$ |
97,903 |
|
||
Total minimum future rental payments under operating leases described above and in aggregate are as follows:
|
Years ended December 31, |
Amount |
||
|
2021 |
$ |
187,077 |
|
|
2022 |
|
191,264 |
|
|
2023 |
|
145,802 |
|
|
Total minimum future payments |
|
524,143 |
|
|
Less: interest |
|
26,662 |
|
|
Present value of operating lease liabilities |
$ |
497,481 |
|
F-21
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note K — Other Uncertainties
The current outbreak of the strain of coronavirus known as COVID-19 has reduced the enrollment of new patients, starting in April 2020. Existing patients continued to receive their treatments, but new patient enrollments were generally placed on hold at most of the Company’s clinical trial sites. Enrollment appears to have returned to more normal levels in March 2021. Under the assumption that patient enrollments will resume planned in the upcoming months, the Company does not expect a significant extension in the duration of its clinical trials. If patient enrollments do not resume to planned levels, the Company may incur additional clinical trial expenses.
Note L — Related Parties
In May 2017, the Company entered into a Provider Services Agreement (“PSA”) with a minority stockholder. In this agreement, the Company provides use of its office space over time periods that match the Company’s initial rental period and annual extensions. The proceeds from the PSA are recorded as a reduction of the Company’s rent expense, salaries and benefits related to work performed, and related overhead costs. In October 2020, this agreement was expanded to include some of the additional space that the Company began to lease in October 2020.
At December 31, 2020 and 2019, the Company was holding a $36,000 deposit related to the PSA. This account is entitled “Related party deposit” on the balance sheet. This deposit will be returned to the minority stockholder at the end of the PSA once all charges have been settled. At December 31, 2020 and 2019, the Company had a receivable of approximately $6,500 and $6,300, respectively, related to this agreement. This receivable is included in “Other current assets” on the balance sheet.
Note M — Income Taxes
The Company recorded research & development credits, net of expenses, of $98,400 and $118,200 in 2020 and 2019, respectively since the Federal and Connecticut tax credits exceeded the tax liabilities. Included in these amounts are Connecticut State tax expense of approximately $12,600 and $12,700 for 2020 and 2019, respectively. The Connecticut taxes are based upon the Company’s equity balances.
The Company recognizes Connecticut tax credits in the years that they are received.
At December 31, 2020, aside from the federal research and development tax credits used to offset Social Security taxes, the Company had federal tax credit carryforwards of approximately $200,000 which are available to offset future taxable income expiring at various times beginning in 2033.
At December 31, 2020, the Company has Connecticut research and development tax credit carryforwards of approximately $204,000 which are available to offset future Connecticut taxable income.
At December 31, 2020, the Company generated Connecticut and Federal net operating loss carryforwards of approximately $20.0 million. For the federal net operating loss carryforwards, approximately $7.0 million expire at various dates beginning in 2033. Under the Tax Cuts and Jobs Act (“TCJA”), passed on December 22, 2017, corporate net operating losses generated beginning in 2018 cannot be carried back but are carried forward indefinitely.
In the accompanying statement of operations, research and development credits are included in other income, and taxes, other than income taxes, are included in general and administrative costs.
These net operating losses could offset only up to 80% of taxable income in future years (pre-2018 net operating losses could continue to offset taxable income with no limitation). The approximate $5.4 million, $4.5 million, and $3.1 million of Federal net operating loss generated in 2020, 2019, and 2018, respectively, is subject to this limitation.
The Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”), enacted and signed into law by President Trump on March 27, 2020 in response to the COVID-19 pandemic, temporarily suspends changes to the net operating loss rules made in the TCJA. The first change is that it temporarily removes the taxable income limitation, allowing net operating loss carryforwards to fully offset income. For tax years beginning after December 31, 2017 and before January 1, 2021, the Company is eligible to offset 100% of taxable income in years prior to January 1, 2021 and 80% of taxable income in years beginning January 1, 2021.
F-22
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
December 31, 2020 and 2019
Note M — Income Taxes (cont.)
Since the ability to use net operating loss carryforwards and credits in the future is uncertain, they are recorded as deferred tax assets with a full valuation allowance. They will continue to be recognized in the years that they are utilized.
The components of the deferred tax assets at December 31, 2020 and 2019 are comprised of:
|
2020 |
2019 |
|||||||
|
Federal net operating carryforward |
$ |
20,068,600 |
|
$ |
14,642,400 |
|
||
|
Share-based compensation |
|
1,246,500 |
|
|
1,008,860 |
|
||
|
Anticipated marginal tax rate |
|
21 |
% |
|
21 |
% |
||
|
|
4,476,200 |
|
|
3,286,800 |
|
|||
|
Federal research and development credit |
|
200,500 |
|
|
200,500 |
|
||
|
Total Federal deferred tax assets |
|
4,676,700 |
|
|
3,487,300 |
|
||
|
Valuation allowance |
|
(4,676,700 |
) |
|
(3,487,300 |
) |
||
|
Net deferred tax asset |
$ |
— |
|
$ |
— |
|
||
|
2020 |
2019 |
|||||||
|
Connecticut net operating carryforward |
$ |
20,013,300 |
|
$ |
14,599,700 |
|
||
|
Anticipated marginal tax rate |
|
7.5 |
% |
|
7.5 |
% |
||
|
|
1,501,000 |
|
|
1,095,000 |
|
|||
|
|
|
|
|
|||||
|
State research and development credit |
|
204,200 |
|
|
136,000 |
|
||
|
Total Connecticut deferred tax assets |
|
1,705,200 |
|
|
1,231,000 |
|
||
|
Valuation allowance |
|
(1,705,200 |
) |
|
(1,231,000 |
) |
||
|
Net deferred tax asset |
$ |
— |
|
$ |
— |
|
||
Note N — Retirement Plan — Defined Contribution
The Company maintains a defined contribution plan for all employees age 21 and older who have completed one month of service. This 401K plan began for payrolls after July 1, 2017. The Company makes a matching contribution equal to 100% of an employee’s contribution, up to 3% of an employee’s eligible earnings. The Company match is vested after one year of service. Retirement expense for this plan was approximately $32,000 and $23,000 in 2020 and 2019, respectively.
Note O — Subsequent Events
The Company evaluated subsequent events for financial reporting purposes through September 20, 2021, the date which the audited financial statements were issued to determine whether any events occurred that required adjustment to or disclosure in the accompanying financial statements. The Company concluded that no additional subsequent events required disclosure in these financial statements other than those disclosed in these notes to the financial statements.
F-23
INTENSITY THERAPEUTICS, INC.
Balance Sheets (unaudited)
|
September 30, |
||||||||
|
2021 |
2020 |
|||||||
|
ASSETS |
|
|
|
|
||||
|
Current assets: |
|
|
|
|
||||
|
Cash and cash equivalents |
$ |
7,408,192 |
|
$ |
3,136,535 |
|
||
|
Investments |
|
— |
|
|
7,898,751 |
|
||
|
Other current assets |
|
289,707 |
|
|
317,595 |
|
||
|
Total current assets |
|
7,697,899 |
|
|
11,352,881 |
|
||
|
Deferred offering costs |
|
31,847 |
|
|
— |
|
||
|
Right-of-use asset, net |
|
361,786 |
|
|
177,832 |
|
||
|
Other assets |
|
17,739 |
|
|
31,138 |
|
||
|
Total assets |
$ |
8,109,271 |
|
$ |
11,561,851 |
|
||
|
LIABILITIES, REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIENCY |
|
|
|
|
||||
|
Current liabilities: |
|
|
|
|
||||
|
Accounts payable |
$ |
295,097 |
|
$ |
255,417 |
|
||
|
Accrued expenses |
|
1,945,069 |
|
|
1,127,444 |
|
||
|
Current lease liability |
|
179,519 |
|
|
168,515 |
|
||
|
Convertible note and accrued interest |
|
2,001,644 |
|
|
— |
|
||
|
Total current liabilities |
|
4,421,329 |
|
|
1,551,376 |
|
||
|
Long-term liabilities: |
|
|
|
|
||||
|
Related party deposit |
|
36,000 |
|
|
36,000 |
|
||
|
Long-term lease liability |
|
190,611 |
|
|
16,075 |
|
||
|
Total long-term liabilities |
|
226,611 |
|
|
52,075 |
|
||
|
Total liabilities |
|
4,647,940 |
|
|
1,603,451 |
|
||
|
Series A redeemable convertible preferred stock, par value $.0001. Authorized, issued, and outstanding shares of 5,000,000 as of September 30, 2021 and 2020. Liquidation preference of $20,000,000 as of September 30, 2021. |
|
10,000,000 |
|
|
10,000,000 |
|
||
|
STOCKHOLDERS’ DEFICIENCY |
|
|
|
|
||||
|
Series B convertible preferred stocks, par value $.0001. Authorized, issued, and outstanding shares of 1,449,113 as of September 30, 2021 and 2020. Liquidation preference of $3,260,504 as of September 30, 2021. |
|
145 |
|
|
145 |
|
||
|
Series C convertible preferred stocks, par value $.0001. Authorized shares of 1,800,606 and 6,000,000 at September 30, 2021 and 2020, respectively. Issued and outstanding shares of 1,800,606 at September 30, 2021 and 2020. Liquidation preference of $4,051,364 as of September 30, 2021. |
|
180 |
|
|
180 |
|
||
|
Common stock, par value $.0001. Authorized shares of 50,000,000 and 30,000,000 at September 30, 2021 and 2020, respectively. Issued and outstanding shares of 6,820,211 at September 30, 2021 and 2020. |
|
682 |
|
|
682 |
|
||
|
Additional paid in capital |
|
22,130,317 |
|
|
21,548,325 |
|
||
|
Note receivable for purchase of common stock: related party |
|
— |
|
|
(50,000 |
) |
||
|
Accumulated deficit |
|
(28,669,993 |
) |
|
(21,540,932 |
) |
||
|
Total stockholders’ deficiency |
|
(6,538,669 |
) |
|
(41,600 |
) |
||
|
Total liabilities, redeemable convertible preferred stock and stockholders’ deficiency |
$ |
8,109,271 |
|
$ |
11,561,851 |
|
||
The accompanying notes are an integral part of these financial statements.
F-24
INTENSITY THERAPEUTICS, INC.
Statements of Operations (unaudited)
|
Nine Months Ended |
||||||||
|
2021 |
2020 |
|||||||
|
Operating expenses: |
|
|
|
|
||||
|
Research and development costs |
$ |
4,418,919 |
|
$ |
3,643,840 |
|
||
|
General and administrative costs |
|
1,184,551 |
|
|
922,095 |
|
||
|
Total operating expenses |
|
5,603,470 |
|
|
4,565,935 |
|
||
|
Loss from operations |
|
(5,603,470 |
) |
|
(4,565,935 |
) |
||
|
Other income: |
|
|
|
|
||||
|
Interest income |
|
2,261 |
|
|
68,028 |
|
||
|
Other |
|
106,727 |
|
|
102,006 |
|
||
|
Net loss |
$ |
(5,494,482 |
) |
$ |
(4,395,901 |
) |
||
|
Loss per share |
|
|
|
|
||||
|
Loss per share, basic and diluted |
$ |
(0.81 |
) |
$ |
(0.64 |
) |
||
|
Weighted average number of shares of common stock, basic and diluted |
|
6,820,211 |
|
|
6,818,631 |
|
||
The accompanying notes are an integral part of these financial statements.
F-25
INTENSITY THERAPEUTICS, INC.
Statements of Changes in Redeemable Convertible Preferred Stock and Stockholders’ Deficiency
Nine Months Ended September 30, 2021 and 2020 (unaudited)
|
Series A |
Series B |
Series C |
Common Stock |
Additional |
Note |
Accumulated Deficit |
Stockholders’ Deficiency |
||||||||||||||||||||||||||||
|
Shares |
Amount |
Shares |
Amount |
Shares |
Amount |
Shares |
Amount |
||||||||||||||||||||||||||||
|
Balances at December 31, 2019 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
695,653 |
$ |
70 |
6,805,994 |
$ |
681 |
$ |
14,843,168 |
$ |
(75,000 |
) |
$ |
(17,145,031 |
) |
$ |
(2,375,967 |
) |
||||||||||||
|
Stock-based compensation expense |
|
|
|
|
|
437,634 |
|
|
|
|
|
437,634 |
|
||||||||||||||||||||||
|
Issuance of Series C convertible preferred stock – net of placement fees of $167,591 |
|
|
1,104,953 |
|
110 |
14,217 |
|
1 |
|
6,267,523 |
|
|
|
|
|
6,267,634 |
|
||||||||||||||||||
|
Repayment of shareholder note |
|
|
|
|
|
|
25,000 |
|
|
|
|
25,000 |
|
||||||||||||||||||||||
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(4,395,901 |
) |
|
(4,395,901 |
) |
||||||||||||
|
Balances at September 30, 2020 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
1,800,606 |
$ |
180 |
6,820,211 |
$ |
682 |
$ |
21,548,325 |
$ |
(50,000 |
) |
$ |
(21,540,932 |
) |
$ |
(41,600 |
) |
||||||||||||
|
Balances at December 31, 2020 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
1,800,606 |
$ |
180 |
6,820,211 |
$ |
682 |
$ |
21,666,178 |
$ |
(50,000 |
) |
$ |
(23,175,511 |
) |
$ |
(1,558,326 |
) |
||||||||||||
|
Stock-based compensation expense |
|
|
|
|
|
464,139 |
|
|
|
|
|
464,139 |
|
||||||||||||||||||||||
|
Repayment of shareholder note |
|
|
|
|
|
|
50,000 |
|
|
|
|
50,000 |
|
||||||||||||||||||||||
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(5,494,482 |
) |
|
(5,494,482 |
) |
||||||||||||
|
Balances at September 30, 2021 |
5,000,000 |
$ |
10,000,000 |
1,449,113 |
$ |
145 |
1,800,606 |
$ |
180 |
6,820,211 |
$ |
682 |
$ |
22,130,317 |
$ |
— |
|
$ |
(28,669,993 |
) |
$ |
(6,538,669 |
) |
||||||||||||
The accompanying notes are an integral part of these financial statements.
F-26
INTENSITY THERAPEUTICS, INC.
Statements of Cash Flows (unaudited)
|
Nine Months Ended |
||||||||
|
2021 |
2020 |
|||||||
|
Cash flows from operating activities: |
|
|
|
|
||||
|
Net loss |
$ |
(5,494,482 |
) |
$ |
(4,395,901 |
) |
||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
||||
|
Reduction in carrying amount of right-of-use asset |
|
128,454 |
|
|
74,836 |
|
||
|
Stock-based compensation expense |
|
464,139 |
|
|
437,634 |
|
||
|
Subtotal of non-cash expenses |
|
592,593 |
|
|
512,470 |
|
||
|
Changes in operating assets and liabilities, net: |
|
|
|
|
||||
|
Other current assets |
|
22,163 |
|
|
(62,021 |
) |
||
|
Deferred offering costs |
|
(31,847 |
) |
|
— |
|
||
|
Other assets |
|
13,964 |
|
|
(1,697 |
) |
||
|
Accounts payable |
|
72,390 |
|
|
(138,546 |
) |
||
|
Accrued expenses |
|
993,026 |
|
|
510,376 |
|
||
|
Accrued interest on convertible note |
|
1,644 |
|
|
— |
|
||
|
Change in lease liabilities |
|
(127,351 |
) |
|
(75,680 |
) |
||
|
Subtotal of changes in operating assets and liabilities |
|
943,989 |
|
|
232,432 |
|
||
|
Net cash used in operating activities |
|
(3,957,900 |
) |
|
(3,650,999 |
) |
||
|
Cash flows from investing activities: |
|
|
|
|
||||
|
Purchase of short term investments |
|
— |
|
|
(10,898,395 |
) |
||
|
Redemptions of short term investments |
|
— |
|
|
7,564,457 |
|
||
|
Net cash used in investing activities |
|
— |
|
|
(3,333,938 |
) |
||
|
Cash flows from financing activities: |
|
|
|
|
||||
|
Payment received under shareholder note |
|
50,000 |
|
|
25,000 |
|
||
|
Proceeds from sale of preferred stock and preferred stock and/or common stock warrants, net |
|
— |
|
|
6,267,634 |
|
||
|
Proceeds from sale of convertible note |
|
2,000,000 |
|
|
— |
|
||
|
Net cash provided by financing activities |
|
2,050,000 |
|
|
6,292,634 |
|
||
|
Net decrease in cash and cash equivalents |
|
(1,907,900 |
) |
|
(692,303) |
|
||
|
Cash and cash equivalents at beginning of year |
|
9,316,092 |
|
|
3,828,838 |
|
||
|
Cash and cash equivalents at end of year |
$ |
7,408,192 |
|
$ |
3,136,535 |
|
||
|
Supplemental disclosure of non-cash financing activities: |
|
|
|
|
||||
|
Common stock issued for services to placement agent. |
|
|
$ |
81,748 |
|
|||
The accompanying notes are an integral part of these financial statements.
F-27
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note A — Nature of Business
[1] Corporate history:
Intensity Therapeutics, Inc. (“the Company”) is a Connecticut based biotechnology company whose treatment approach addresses both the regional and systemic nature of a patient’s cancer. The Company’s DfuseRxSM technology platform has identified a lead drug, INT230-6.
[2] Propriety products and technology portfolios:
The Company’s Phase 1/2 protocol has been authorized to proceed by both the United States Food & Drug Administration (“FDA”) and Health Canada for INT230-6. In May 2017, the Company began the clinical study.
In April 2019, the FDA granted Fast Track designation to the Company’s development program evaluating INT230-6 for the treatment of patients with relapsed or metastatic triple negative breast cancer who have failed at least two prior lines of therapy.
In June 2019, the Company entered into an agreement with Merck (known as MSD outside the United States and Canada), through a subsidiary of Merck, to evaluate the combination of the Company’s lead product candidate INT230-6 and KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 (programmed death receptor-1) therapy, in patients with advanced solid malignancies including pancreatic, bile duct, squamous cell and non-MSI high colon cancers.
In April 2020, the Company entered into a clinical trial collaboration agreement with Bristol Myers Squibb (NYSE: BMY) Company to evaluate the safety and efficacy of our INT230-6 with BMY’s Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) immune checkpoint inhibitor Yervoy® (ipilimumab). The combination will be evaluated in patients with breast cancer, liver cancer and advanced sarcoma.
The Company is now in the Phase 2 of the clinical trial. Since the KEYTRUDA injections may continue for up to two years from the initial dose, the Company anticipates that Phase 1/2 drug testing could continue into 2024.
In March 2021, the Company began INVINCIBLE study (IT-02) which is a Phase 2 Randomized, Window of Opportunity Trial in Early Stage Breast Cancer. The Company anticipates this study to be completed in 2022.
Note B — Liquidity and Plan of Operation
The accompanying financial statements have been prepared in conformity with generally accepted accounting principles, which contemplate continuation of the Company as a going concern.
The Company is a development stage company and has not generated any revenue from its product candidates. The Company, therefore, has experienced net losses and negative cash flows from operations each year since its inception. Through September 30, 2021, the Company has an accumulated deficit of approximately $28.7 million. The Company’s operations have been financed primarily through the sale of equity securities. The Company’s net loss for the nine months ended September 30, 2021 and 2020 were approximately $5.5 million and $4.4 million, respectively.
To date, the Company has not obtained regulatory approval for any of its product candidates. The Company expects to incur significant expenses to complete development of its product candidates. The Company may never be able to obtain regulatory approval for the marketing of any of its product candidates in the United States or internationally and there can be no assurance that the Company will generate revenues or ever achieve profitability. The Company does not expect to receive significant product revenue in the near term. The Company, therefore, expects to continue to incur substantial losses for the foreseeable future.
Cash and cash equivalents at September 30, 2021 totaled approximately $7.4 million. Until such time, if ever, as the Company can generate substantial product revenue, the Company expects to finance its cash needs through a combination of equity offerings and debt financings. The Company does not have any committed external source of funds. To the extent that the Company raises additional capital through the sale of equity or convertible debt securities, the ownership interest of the Company stockholders will be diluted, and the terms of these securities may include
F-28
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note B — Liquidity and Plan of Operation (cont.)
liquidation or other preferences that adversely affect the rights of a common stockholder. If the Company is unable to raise additional funds through equity or debt financings when needed, the Company may be required to delay, limit, reduce or terminate its research and product development.
Based on cash on hand at September 30, 2021, we believe that the Company will have sufficient cash to fund planned operations through September 30, 2022. However, the acceleration or reduction of cash outflows by management can significantly impact the timing for raising additional capital to complete development of its product candidates. To continue development, the Company will need to raise additional capital through debt and/or equity financing. Additional capital may not be available on terms favorable to the Company, if at all. If the Company does not raise at least $10,000,000 in an Initial Public Offering, two-thirds of the Series A Redeemable Convertible Preferred Shareholders may vote any time after May 18, 2022 to redeem their shares at a total amount of $10,000,000. The Company does not know if its future offerings will succeed. Accordingly, no assurances can be given that management will be successful in these endeavors. The Company’s recurring losses from operations have caused management to determine there is substantial doubt about the Company’s ability to continue as a going concern. These Financial Statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities or any other adjustments that might be necessary should the Company be unable to continue as a going concern.
Note C — Summary of Significant Accounting Policies and Accounts
[1] Basis of presentation:
The accompanying financial statements include the accounts of Intensity Therapeutics, Inc. The Company neither owns nor controls any subsidiary companies. The accompanying financial statements have been prepared by the Company in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and reflect the operations of the Company.
[2] Use of estimates:
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Certain accounting principles require subjective and complex judgments to be used in the preparation of financial statements. Accordingly, a different financial presentation could result depending on the judgments, estimates, or assumptions that are used.
The Company utilizes significant estimates and assumptions in valuing its stock-based compensation awards. An additional significant estimate is that these financial statements are based on the assumption of the Company continuing as a going concern. See note B with regards to the Company’s ability to continue as a going concern.
[3] Concentration of credit risk:
The Company’s financial instruments that are exposed to concentrations of credit risk consist entirely of cash, certificates of deposit, and investments in U.S. Treasury bills. These financial instruments are held at two major U.S. financial institutions. The cash accounts are insured by the Federal Deposit Insurance Corporation (“FDIC”) up to regulatory limits. At all times throughout the nine months ended September 30, 2021 and 2020, the Company’s cash balances exceeded the FDIC insurance limit. The Company has not experienced any losses in such accounts. The investments in U.S. Treasury bills are not FDIC insured, but are backed by the U.S. government. U.S. Treasury bills are subject to market risk if they are sold prior to maturity.
F-29
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
[4] Cash and cash equivalents:
The Company considers all liquid investments with an original maturity of three months or less to be cash equivalents.
[5] Fair value measurement:
The Company reports its investments at fair value. Fair value is an estimate of the exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants (i.e., the exit price at the measurement date). Fair value measurements are not adjusted for transaction costs. A fair value hierarchy provides for prioritizing inputs to valuation techniques used to measure fair value into three levels:
Level 1 Unadjusted quoted prices in active markets for identical assets or liabilities.
Level 2 Inputs other than quoted market prices that are observable, either directly or indirectly, and reasonably available. Observable inputs reflect the assumptions market participants would use in pricing the asset or liability and are developed based on market data obtained from sources independent of the Company.
Level 3 Unobservable inputs. Unobservable inputs reflect the assumptions that the Company develops based on available information about what market participants would use in valuing the asset or liability.
An asset’s or liability’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Availability of observable inputs can vary and is affected by a variety of factors. The Company uses judgment in determining fair value of assets and liabilities and Level 3 assets and liabilities involve greater judgment than Level 1 or Level 2 assets or liabilities.
At September 30, 2020, the Company had investments of $2,900,000 in a bank certificates of deposit. U.S. government securities are valued using a model that incorporates market observable data, such as reported sales of similar securities, broker quotes, yields, bids, offers, and reference data. U.S. Treasury Bills are categorized in Level 1 of the fair value hierarchy. At September 30, 2020 the Company had investments of approximately $5,000,000 in US Treasury Bills. U.S. government securities are valued using a model that incorporates market observable data, such as reported sales of similar securities, broker quotes, yields, bids, offers, and reference data.
The Company’s financial instruments, including cash equivalents, investments and current liabilities are carried at cost, which approximates fair value due to the short-term nature of these instruments.
[6] Stock-based compensation:
The Company accounts for stock-based compensation to employees and non-employees in conformity with the provisions of Accounting Standards Codification (“ASC”) ASC Topic 718, “Compensation — Stock Compensation”. Stock compensation to employees consist of stock option grants that were recognized in the statements of operations based on their fair values at the date of grant.
The Company calculates the fair value of option grants utilizing the Black-Scholes pricing model. The amount of stock-based compensation recognized during a period is based on the value of the portion of the awards that are ultimately expected to vest. Forfeitures are recognized as they occur.
F-30
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
The resulting stock-based compensation expense for both employee and non-employee awards is generally recognized on a straight-line basis over the requisite service period of the award.
[7] Research and development and patent costs:
Research and development costs are charged to operations as they are incurred. Legal fees and other direct costs incurred in obtaining and protecting patents are also expensed as incurred, due to the uncertainty with respect to future cash flows resulting from the patents and are included as part of general and administrative expenses in the Company’s Statements of Operations.
[8] Income taxes:
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. ASC 740 prescribes the use of the asset-and-liability method whereby deferred tax assets and liabilities are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company utilizes a valuation allowance to reduce deferred tax assets to their estimated realizable value.
The Company accounts for uncertain tax positions in accordance with the provisions of ASC 740. When uncertain tax positions exist, the Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely than not be realized.
The determination as to whether the tax benefit will more likely than not be realized is based upon the technical merits of the tax position as well as consideration of the available facts and circumstances. At
September 30, 2021 and 2020, the Company does not have any significant uncertain tax positions.
There are no estimated interest costs and penalties provided for in the Company’s financial statements for the nine months ended September 30, 2021 and 2020. If at any time the Company should record interest and penalties in connection with an uncertain tax position, the interest and penalties will be expensed within the income tax line.
The Company’s income tax returns are subject to Federal, state and local income tax examination by the authorities for the last three tax years.
[9] Stock issuance costs:
For the nine months ended September 30, 2020, the Company had incurred approximately $167,600 of costs related to the sale of the Series C preferred stock. These costs were recorded as a deduction to Additional Paid in Capital.
[10] Leases:
The Company determines if an arrangement contains a lease at contract inception. With the exception of short-term leases (leases with terms less than 12 months), all leases with contractual fixed costs are recorded on the balance sheet on the commencement date as a right-of-use (ROU) asset and a lease liability. Lease liabilities to be paid over the next twelve months are classified as current lease liability and all other lease obligations are classified as Long-term lease liability. Lease liabilities are initially measured at the present value of the future minimum lease payments and subsequently increased to reflect the interest accrued and reduced by the lease payments made. The Company’s building leases require a pro-rata share of operating expense and real estate taxes, which are variable in nature and excluded from the measurement of lease liabilities. ROU assets are initially measured at the present value of the future minimum lease payments adjusted for any prior lease pre-payments, lease incentives and initial direct costs. Certain leases contain escalation, renewal and/or termination options that are factored into the ROU asset as appropriate. Operating leases result in a straight-line rent expense over the expected lease term.
F-31
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
The Company uses its estimated incremental borrowing rate, which is derived from information available at the lease commencement date, in determining the present value of future lease payments, if the rate implicit in the lease is not readily determinable. Consideration is given to publicly available data for instruments with similar characteristics when calculating incremental borrowing rates. This incremental borrowing rate estimate is based on a synthetic credit rating derived from the Company’s market capitalization, the treasury yield curve, and corporate yield spreads.
[11] Basic and dilutive loss per share:
Basic net loss per share is determined using the weighted average number of shares of common stock outstanding during each period. Dilutive net loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as convertible preferred stock, stock options, and stock warrants, which would result in the issuance of incremental shares of common stock. The computation of diluted net loss per share does not include the conversion of securities that would have an anti-dilutive effect. Potential common shares issuable upon conversion of preferred stock, exercise of stock options, and exercise of warrants that are excluded from the computation of diluted weighted average shares outstanding are 10,718,719 and 10,230,719 as of September 30, 2021 and 2020, respectively. The basic and diluted computation of net loss per share for the Company are the same because the effects of the Company’s convertible securities would be anti-dilutive. All common and preferred stock participate equally in dividends and the distribution of earnings. For purposes of computing earnings per share, all series of preferred stock are considered participating securities. Therefore, the Company must calculate basic and diluted earnings per share using the two-class method. Under the two-class method, net income for the period is allocated between common stockholders and participating securities according to dividends declared and participation rights in undistributed earnings. As the preferred shareholders have no obligation to fund losses no portion of net loss was allocated to the participating securities for the periods ended September 30, 2021 and 2020.
The following are shares excluded from the computation of diluted weighted average shares outstanding at September 30, 2021 and 2020:
|
September 30, |
September 30, |
|||
|
Preferred stock Series A issued |
5,000,000 |
5,000,000 |
||
|
Preferred stock Series B issued |
1,449,113 |
1,449,113 |
||
|
Preferred stock Series C issued |
1,800,606 |
1,800,606 |
||
|
Options outstanding |
1,822,500 |
1,706,500 |
||
|
Warrants outstanding |
646,500 |
274,500 |
||
|
10,718,719 |
10,230,719 |
[12] Recent accounting pronouncements:
In June 2018, the FASB issued ASU No. 2018-07, “Compensation — Stock Compensation: Improvements to Nonemployee Shared-Based Payment Accounting” (“Topic 718”), which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718 to include share-based payments transactions for acquiring goods and services from nonemployees. Some of the areas for simplification apply only to nonpublic entities.
The amendments specify that Topic 718 applies to all share-based payment transactions in which a grantor acquires goods or services to be used or consumed in a grantor’s own operations by issuing share-based payment awards. The amendments also clarify that Topic 718 does not apply to share-based payments used to effectively provide (1) financing to the issuer or (2) awards granted in conjunction with selling goods or services to customers as part of a contract accounted for under Topic 606, “Revenue from Contracts with Customers.” The amendments in this update are effective for nonpublic companies for fiscal years beginning after December 15, 2019. Early adoption is permitted, but no earlier than a Company’s adoption date of Topic 606. The Company has adopted this pronouncement as of January 1, 2020, noting no material impact on the Company’s financial statements.
F-32
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note C — Summary of Significant Accounting Policies and Accounts (cont.)
[13] Deferred offering costs:
Deferred offering costs consist of underwriting, legal, accounting and other expenses that are directly related to the proposed initial offering and that will be charged to stockholders’ equity upon the completion of the initial proposed offering. Should the initial proposed offering prove to be unsuccessful, these deferred costs, as well as additional expenses incurred, will be charged to operations.
Note D — Other Current Assets
Other current assets at September 30 include:
|
2021 |
2020 |
|||||
|
Advance to subcontractor |
$ |
150,000 |
$ |
150,000 |
||
|
Prepaid insurance |
|
21,823 |
|
63,929 |
||
|
Taxes receivable |
|
47,223 |
|
37,061 |
||
|
Other current assets |
|
70,661 |
|
66,605 |
||
|
$ |
289,707 |
$ |
317,595 |
|||
The $150,000 advance is to a company which provides services in managing the Company’s clinical trials. The advance is intended to offset the normal delay in the Company reimbursing them for their services and for their payments to the Company’s clinical sites.
Note E — Note Receivable
On February 4, 2015, the Company received a $75,000 note from its Chief Medical Officer. The note was used to fund 75% of a common stock purchase. Interest accrues at the rate of three percent per annum, compounding annually on each anniversary of the date of the note creation.
In 2019, the payment terms were amended to the following schedule, unless paid earlier:
• $25,000 due on or before July 11, 2020;
• $25,000 due on or before July 11, 2021; and
• the remaining balance, including the interest due, in full on or before February 4, 2022.
Sale of the related securities would require a repayment of the note. The note is recorded as a reduction to stockholders’ equity since the note was used to purchase the Company’s common stock. Accrued interest on this note was approximately $13,000 at September 30, 2020, and is included in other assets and interest income.
The balance due on this note was $50,000 at September 30, 2020. In July 2021 the Chief Medical Officer made a payment of $25,000. In August 2021, the Board approved a bonus to the Chief Medical Officer for $25,000, which was the remaining balance at that time, and the $15,073 of related accrued interest in order to extinguish the remaining balance of his note.
F-33
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note F — Accrued Expenses
Accrued expenses at September 30 include:
|
2021 |
2020 |
|||||
|
Accrued vacation, wages, and related payroll taxes |
$ |
270,487 |
$ |
236,655 |
||
|
Patient costs incurred but not yet invoiced |
|
1,665,311 |
|
881,517 |
||
|
Accrued other |
|
9,271 |
|
9,272 |
||
|
$ |
1,945,069 |
$ |
1,127,444 |
|||
Note G — Convertible Note
On September 20, 2021, the Company entered into convertible debt agreement (the “2021 Convertible Note”) with a holder for aggregate principal of $2,000,000 due September 20, 2023, at the following conversion terms. The outstanding principal balance together with the unpaid and accrued interest of the notes will be automatically converted upon the earlier of (i) an Initial Public Offering (IPO) in excess of $10,000,000 gross proceeds, (ii) a sale event of all or substantially all of the company’s assets or a majority of its equity securities, (iii) Non IPO financing by selling preferred stock in an equity offering other than an IPO or (iv) maturity date of September 20, 2023. If an IPO, sale event or Non IPO financing occurs between September 20, 2021 through September 19, 2022 a conversion price discount of 25% would be assessed, if between September 20, 2022 through March 19, 2023 a conversion price discount of 30% would be assessed, if between March 20, 2023 through September 20, 2023 a conversion price discount of 35% would be assessed. Otherwise at the maturity date a conversion price of $7.50 per share would be assessed. The 2021 Convertible Note accrues interest at 3% per annum, convertible to shares as previously described herein. The occurrence of any of the following shall constitute an Event of Default: a) Failure to pay when due any principal payment; b) voluntary bankruptcy or insolvency proceedings; c) involuntary bankruptcy or insolvency proceedings; d) judgements in excess of $500,000; or e) defaults under other indebtedness. Under these occurrences, the holder may declare all outstanding principal and interest payable to be immediately due and payable.
The balance at September 30, 2021 consists of the $2,000,000 purchase price plus $1,644 of accrued interest through September 30, 2021, which totals the $2,001,644 that appears on the balance sheet.
This convertible note has a contingent beneficial conversion feature. The value of this beneficial conversion feature has not yet been determined since an Initial Public Offering price has not been determined. Once the intrinsic value of the beneficial conversion feature is determined it will be charged to interest expense over the period from when the amount was determined to the time the note becomes convertible into common stock.
Note H — Stockholders’ Equity
[1] Authorized and outstanding shares:
The total number of shares of all classes of stock which the Company shall have authority to issue, at September 30, in which all shares have a par value of $0.0001 are as follows:
|
2021 |
2020 |
|||
|
Common stock |
50,000,000 |
30,000,000 |
||
|
Preferred stock: |
||||
|
Series A |
5,000,000 |
5,000,000 |
||
|
Series B |
1,449,113 |
1,449,113 |
||
|
Series C |
1,800,606 |
6,000,000 |
||
|
undesignated |
11,750,281 |
5,050,887 |
||
|
Total Preferred stock |
20,000,000 |
17,500,000 |
F-34
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note H — Stockholders’ Equity (cont.)
The Company has issued the following number of shares, options, and warrants at September 30:
|
2021 |
2020 |
|||
|
Common stock issued |
6,820,211 |
6,820,211 |
||
|
Preferred stock Series A issued |
5,000,000 |
5,000,000 |
||
|
Preferred stock Series B issued |
1,449,113 |
1,449,113 |
||
|
Preferred stock Series C issued |
1,800,606 |
1,800,606 |
||
|
Total outstanding shares |
15,069,930 |
15,069,930 |
||
|
Options outstanding |
1,822,500 |
1,706,500 |
||
|
Warrants outstanding |
646,500 |
274,500 |
||
|
Total outstanding shares, options and warrants |
17,538,930 |
17,050,930 |
In 2020, the Company amended its 2013 Stock Option Plan to increase the number of authorized shares available under the plan from 1,800,000 to 4,500,000. At September 30, 2021, there are 2,677,500 unissued options reserved for issuance under the Company’s 2013 Stock Option Plan.
In 2019, the Company initiated an offering of Series C Preferred Stock (“Series C”) with a par value of $0.0001, in which the Company was authorized to sell up to 6 million shares. The Company sold 1,800,606 shares (1,104,953 in 2020 and 695,653 in 2019) for net proceeds of $10,044,545 ($6,185,885 in 2020 and $3,858,660 in 2019.) All of the sales in 2020 occurred in the first six months of 2020. In conjunction with the Series C raise, the Company issued warrants, which the holder can convert into 60,000 shares of Series C preferred stock or common stock. Each share of the preferred stock or common stock under the warrant is converted for $6.25 per share of preferred or common stock. The warrants expire on December 31, 2022. Subsequent to this sale, the number of authorized shares was reduced to 1,800,606. These warrants are not included in the computation of weighted average number of common stock, basic and diluted. These warrants, if exercised, will participate equally in dividends and distributions of earnings.
Through September 30, 2021, the Company has raised approximately:
|
Sale of common stock (2012 to 2020) |
$ |
3.2 million |
|
|
Sale of Series A preferred stock (2015 to 2016) |
|
10.0 million |
|
|
Sale of Series B preferred stock (2018) |
|
6.5 million |
|
|
Sale of Series C preferred stock (2019 to 2020) |
|
10.4 million |
|
|
Total proceeds from the issuance of stock |
$ |
30.1 million |
[2] Voting:
As long as there are issued and outstanding shares of 2,500,000 or more Preferred Stock, the holders of record of the outstanding shares of Preferred Stock shall be entitled to elect two directors of the Company and the holders of record of the shares of Common Stock shall be entitled to elect three directors of the Company.
There are currently four directors of the Company, and the Board of Directors shall be comprised of no more than five directors.
The holders of the Common Stock are entitled to one vote for each share of Common Stock held at all meetings of stockholders. The holders of the Preferred Stock are entitled to cast the number of votes equal to the number of whole shares of Common Stock into which the shares of Preferred Stock held by such holder are convertible as of the record date at a 1:1 conversation ration, subject to adjustments for stock dividends, splits, combinations, and similar events. Preferred holders vote together with common stockholders as of a single class.
F-35
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note H — Stockholders’ Equity (cont.)
[3] Dividends:
The holders of the Preferred Stock are entitled to the same cash dividend that is paid to holders of Common Stock. The Company has not declared or paid any dividends.
[4] Redemption rights:
At any time on or after May 18, 2022, the holders of at least two-thirds of the then outstanding shares of Series A Preferred Stock may elect to cause the Company to redeem all but not less than all of the shares of Series A Preferred Stock at a redemption price per share in cash equal to $2, the Series A Original Issue Price per Share. Series B and C preferred stockholders do not have any redemption rights.
[5] Liquidation preference:
In the event of any voluntary or involuntary liquidation, the holders of the Series A Preferred Stock shall be entitled to be paid out of the assets of the Corporation before any payment shall be made to the holders of Common Stock, in an amount equal to the greater of (i) two times the Series A Original Issue Price, plus any dividends declared but unpaid thereon, or (ii) such amount per share as would have been payable had all shares of Series A Preferred Stock been converted into Common Stock.
The holders of the Series B and C Preferred Stock shall receive distributions pari passu to holders of Series A stockholders. The holders of Series B and C Preferred Stock shall receive the greater of: (i) in preference to any distribution to holders of Common Stock, $2.25 per share of Series B holders and Series C holders, plus declared and unpaid dividends, if any, on each share of Series B and C Preferred Stock, with the balance of any proceeds then distributed pro rata to holders of Company Stock; or (ii) an amount equal to a pro rata share of the proceeds available for distribution to all holders of Company stock (treating the Series B and C Preferred Stock on an as converted to Common Stock basis).
[6] Conversion:
Each share of Series A, B and C Preferred Stock shall be convertible, at the option of the holder thereof, at any time and from time to time, and without the payment of additional consideration by the holder thereof, into such number of fully paid and non-assessable shares of Common Stock as is determined by dividing the Series Original Issue Price by the Series A, B or C Conversion Price (as defined below) in effect at the time of conversion. The “Series A Conversion Price”, “Series B Conversion Price” and “Series C Conversion Price” shall initially be equal to $2.00, $4.50 and $5.75, respectively. Such initial Conversion Price, and the rate at which shares of Preferred Stock may be converted into shares of Common Stock, shall be subject to adjustments as defined in the Certificate of Incorporation.
Upon either (a) the closing of the sale of shares of Common Stock to the public in a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act of 1933, as amended, resulting in at least $15,000,000 of gross proceeds to the Corporation or (b) the date and time, or the occurrence of an event, specified by vote or written consent of the holders of at least a majority of the then outstanding shares of either Series A, B or C Preferred Stock, then (i) all outstanding shares of either Series A, B or C Preferred Stock shall automatically be converted into shares of Common Stock, at the then effective conversion rate as calculated above, and (ii) such shares may not be reissued by the Corporation.
Note I — Common Stock Warrants
The Company had 646,500 and 274,500 outstanding warrants at September 30, 2021 and 2020, respectively. The weighted average exercise price of warrants outstanding at September 30, 2021 and 2020 was $3.00 and $3.36, respectively. Warrants exercisable at September 30, 2021 and 2020 were 568,974 and 243,000, respectively.
F-36
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note I — Common Stock Warrants (cont.)
The following table summarizes information about common stock warrants outstanding and exercisable at September 30, 2021 and 2020:
|
Number of |
Weighted |
||||
|
January 1, 2020 |
243,000 |
$ |
3.05 |
||
|
Issued |
31,500 |
|
5.75 |
||
|
September 30, 2020 |
274,500 |
|
3.36 |
||
|
Exercisable September 30, 2020 |
243,000 |
|
2.49 |
||
|
|
|||||
|
January 1, 2021 |
586,500 |
|
2.72 |
||
|
Issued |
60,000 |
|
5.75 |
||
|
September 30, 2021 |
646,500 |
$ |
3.00 |
||
|
Exercisable September 30, 2021 |
568,974 |
$ |
2.68 |
||
The warrants issued during December 2019 relate to the issuance of the Series C Preferred Stock and were valued at approximately $141,000 and vested immediately with an exercise price of $6.25 and expires in 3 years.
Prior to 2020, the Company had issued 312,000 options to independent contractors. In the fourth quarter of 2020, these options were changed to warrants with the same terms and conditions as the original options.
The aggregate intrinsic value of warrants outstanding was $1,805,000 and $686,250 at September 30, 2021 and 2020, respectively.
The following table summarizes the assumptions used to estimate the fair value of common stock warrants granted during the nine months ended September 30, 2021 and 2020:
|
2021 |
2020 |
|||||
|
Stock price |
$ |
5.75 |
$ |
5.75 |
||
|
Exercise price |
$ |
5.75 |
$ |
5.75 |
||
|
Expected volatility |
|
86.26% – 93.51% |
|
66.60% |
||
|
Risk free interest rates |
|
1.06% – 1.09% |
|
0.55% |
||
|
Expected term |
|
10.00 years |
|
10.00 years |
||
For 2021 and 2020, a dividend yield of 0% was used because the Company has not historically paid, and does not intend to pay, a dividend on common stock in the foreseeable future. The expected stock price volatility was estimated based on the historical volatilities for industry peers, as the Company has no active market for its stock, and limited history for issuance price of its stock.
The risk free rate assumption is determined using the yield currently available on U.S. Treasury zero coupon issues with a remaining term commensurate with the expected term of the award. The expected term of the option represents the period the warrants are expected to be outstanding.
F-37
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note I — Common Stock Warrants (cont.)
The following table summarizes information about common stock warrants outstanding at September 30, 2021:
|
Exercise Price |
Number |
Weighted |
Number |
Weighted |
||||
|
$1.00 – $1.50 |
120,000 |
2.7 years |
120,000 |
2.7 years |
||||
|
$2.00 – $2.50 |
336,500 |
4.2 years |
332,750 |
4.2 years |
||||
|
$4.00 – $4.50 |
38,500 |
7.2 years |
29,124 |
7.1 years |
||||
|
$5.75 – $6.25 |
151,500 |
6.2 years |
87,100 |
3.8 years |
||||
|
646,500 |
568,974 |
The following table summarizes information about common stock warrants outstanding at September 30, 2020:
|
Exercise Price |
Number |
Weighted |
Number Exercisable |
Weighted |
||||
|
$1.00 – $1.50 |
42,500 |
4.3 years |
42,500 |
4.3 years |
||||
|
$2.00 – $2.50 |
140,500 |
5.5 years |
140,500 |
5.5 years |
||||
|
$5.75 – $6.25 |
91,500 |
4.9 years |
60,000 |
2.3 years |
||||
|
274,500 |
243,000 |
Note J — Stock-Based Compensation
The Company has a stock option plan, the 2013 Stock Option Plan (the “Plan”), which is administered by the Committee. Under the Plan, stock options to purchase a total of 4,500,000 shares of common stock, may be granted to eligible employees, officers, directors, and consultants of the Company.
The Company had outstanding options as follows:
|
Number of |
Weighted |
||||
|
January 1, 2020 |
1,347,000 |
$ |
2.93 |
||
|
Issued |
359,500 |
|
5.75 |
||
|
September 30, 2020 |
1,706,500 |
$ |
3.53 |
||
|
Exercisable |
1,000,500 |
$ |
2.47 |
||
|
January 1, 2021 |
1,394,500 |
$ |
3.83 |
||
|
Issued |
428,000 |
|
5.75 |
||
|
September 30, 2021 |
1,822,500 |
$ |
4.28 |
||
|
Exercisable September 30, 2021 |
1,152,250 |
$ |
3.52 |
||
The aggregate intrinsic value of options was $2,655,000 and $3,792,500 at September 30, 2021 and 2020, respectively.
F-38
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note J — Stock-Based Compensation (cont.)
The following table summarizes information about stock options at September 30, 2021:
|
Exercise Price |
Number |
Weighted |
Number |
Weighted |
||||
|
$1.00 – $1.50 |
200,000 |
2.8 years |
200,000 |
2.8 years |
||||
|
$2.00 |
290,000 |
4.7 years |
290,000 |
4.7 years |
||||
|
$4.00 – $4.50 |
545,000 |
7.2 years |
473,750 |
7.2 years |
||||
|
$5.75 |
787,500 |
9.4 years |
188,500 |
9.3 years |
||||
|
1,822,500 |
1,152,250 |
The following table summarizes information about stock options at September 30, 2020:
|
Exercise Price |
Number |
Weighted |
Number Exercisable |
Weighted |
||||
|
$1.00 – $1.50 |
277,500 |
3.7 years |
277,500 |
3.7 years |
||||
|
$2.00 – $2.50 |
486,000 |
5.4 years |
467,250 |
5.4 years |
||||
|
$4.00 – $4.50 |
583,500 |
8.2 years |
255,750 |
8.1 years |
||||
|
$5.75 |
359,500 |
9.9 years |
— |
— years |
||||
|
1,706,500 |
1,000,500 |
Employee option vesting is based on the employee’s continued employment with the Company.
In 2018, the Board of Directors amended the 2013 Stock Option Plan to provide an immediate vesting of outstanding options in the event of a change of control, such as an acquisition, notwithstanding any other provision of the Stock Option Plan.
All options are granted with an exercise price equal to the current fair market value of the stock
At September 30, 2021, total unrecognized compensation cost related to non-vested options was approximately $2,459,000 and is expected to be recognized over the remaining weighted average service period of 3.0 years. The Company recorded stock-based compensation related to stock options of approximately $332,000 and $341,000 with research and development costs for the nine months ended September 30, 2021 and 2020, respectively. The Company recorded stock-based compensation expense related to stock options of approximately $132,000 and $97,000 with general and administrative costs for the nine months ended September 30, 2021 and 2020, respectively.
All options expire ten years from date of grant. Options outstanding begin to expire in August 2024. Options that were granted to employees have vesting periods that vary by award to recipient and range from immediate vesting to a period of up to 4 years.
The shares of stock underlying stock options are restricted securities under U. S. Federal and applicable state securities laws and, as such, may not be transferred, sold, or otherwise disposed of in the United States, except as permitted under U.S. Federal and state securities laws, pursuant to registration or exemption therefrom.
F-39
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note K — Leases
In January 2017, the Company entered a lease for 2,534 square feet of office space at its current location. The lease commenced in May 2017. The initial lease term was two years. In November 2018, the Company exercised the option to extend for an additional three years.
In July 2020, the Company amended this lease to increase office space by an additional 1,653 square feet in the same building. The amended lease that includes the space included in the original lease has monthly rent as follows:
|
Year 1 (October 2020 through September 2021) |
$15,502.50 per month ($44.43 per square foot) |
|
|
Year 2 (October 2021 through September 2022) |
$15,851.42 per month ($45.43 per square foot) |
|
|
Year 3 (October 2022 through September 2023) |
$16,200.33 per month ($46.43 per square foot) |
The Company has an option to extend this amended lease for an additional 3 years at the following amounts:
|
Year 4: (October 2023 through September 2024) |
$16,338.08 per month ($46.83 per square foot) |
|
|
Year 5: (October 2024 through September 2025) |
$16,475.83 per month ($47.22 per square foot) |
|
|
Year 6: (October 2025 through September 2026) |
$16,824.75 per month ($48.22 per square foot) |
The Company has until October 1, 2023 to exercise this option to extend.
The Company also pays a pro-rata share of operating expenses and real estate taxes.
The following table summarizes the balance sheet classification of the operating lease assets and related lease liabilities at September 30:
|
2021 |
2020 |
|||||
|
Operating lease right-of-use assets |
$ |
361,786 |
$ |
177,832 |
||
|
Current portion of operating lease liabilities |
$ |
179,519 |
$ |
168,515 |
||
|
Long-term operating lease liabilities |
|
190,611 |
|
16,075 |
||
|
$ |
370,130 |
$ |
184,590 |
|||
The following variables were used to determine the right-of-use asset and the operating lease liabilities at September 30:
|
2021 |
2020 |
|||
|
Weighted average remaining operating lease term |
2.25 years |
1.92 years |
||
|
Weighted average operating lease discount rate |
3.92% |
5.38% |
Facilities and rent expense for the six months ended September 30 are as follows:
|
2021 |
2020 |
|||||||
|
Operating lease cost |
$ |
140,976 |
|
$ |
83,626 |
|
||
|
Sublease income from related party |
|
(42,136 |
) |
|
(31,383 |
) |
||
|
Net lease cost |
|
98,840 |
|
|
52,243 |
|
||
|
Utilities |
|
9,063 |
|
|
5,045 |
|
||
|
Cleaning, repairs, and other |
|
9,688 |
|
|
7,378 |
|
||
|
Facilities and rent expense |
$ |
117,591 |
|
$ |
64,666 |
|
||
F-40
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note K — Leases (cont.)
Listed below is a summary of future minimum rental payments:
|
October 2021 – December 2021 |
$ |
47,554 |
|
|
January 2022 – December 2022 |
|
191,264 |
|
|
January 2023 – September 2023 |
|
145,803 |
|
|
Total minimum future payments |
|
384,621 |
|
|
Less interest |
|
14,491 |
|
|
Present value of operating lease liabilities |
$ |
370,130 |
Note L — Other Uncertainties
The current outbreak of the strain of coronavirus known as COVID-19 has reduced the enrollment of new patients, starting in April 2020. Existing patients continued to receive their treatments, but new patient enrollments were generally placed on hold at most of the Company’s clinical trial sites. Enrollment appears to have returned to more normal levels in March 2021. Under the assumption that patient enrollments will resume planned in the upcoming months, the Company does not expect a significant extension in the duration of its clinical trials. If patient enrollments do not resume to planned levels, the Company may incur additional clinical trial expenses.
Note M — Related Parties
In May 2017, the Company entered into a Provider Services Agreement (“PSA”) with a minority stockholder. In this agreement, the Company provides use of its office space over time periods that match the Company’s initial rental period and annual extensions. The proceeds from the PSA are recorded as a reduction of the Company’s rent expense, salaries and benefits related to work performed, and related overhead costs. In October 2020, this agreement was expanded to include some of the additional space that the Company began to lease in October 2020.
At September 30, 2021 and 2020, the Company was holding a $36,000 deposit related to the PSA. This account is entitled “Related party deposit” on the balance sheet. This deposit will be returned to the minority stockholder at the end of the PSA once all charges have been settled. At September 30, 2021 and 2020, the Company had a receivable of approximately $0 and $5,300, respectively, related to this agreement. This receivable is included in “Other current assets” on the balance sheet.
Note N — Income Taxes
The Company recorded Federal and Connecticut research and development credits of approximately $106,700 and $102,000 for the nine months ended September 30, 2021 and 2020, respectively. These amounts are included in Other income in the Statements of Operations. Included in general and administrative expense in the Statements of Operations is Delaware franchise tax and Connecticut state income tax expense of approximately $47,800 and $12,800 for the nine months ended September 30, 2021 and 2020, respectively. The Connecticut income taxes are based upon the Company’s equity balances.
The Company recognizes Connecticut tax credits in the years that they are received.
At September 30, 2021, aside from the federal research and development tax credits used to offset Social Security taxes, the Company had federal tax credit carryforwards of approximately $200,000 which are available to offset future taxable income. These credits expire at various times beginning in 2033.
At September 30, 2021, the Company has Connecticut research and development tax credit carryforwards of approximately $204,000 which are available to offset future Connecticut taxable income.
F-41
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note N — Income Taxes (cont.)
At December 31, 2020, the Company generated Connecticut and Federal net operating loss carryforwards of approximately $20.0 million. For the federal net operating loss carryforwards, approximately $7.0 million expire at various dates beginning in 2033. Under the Tax Cuts and Jobs Act (“TCJA”), passed on December 22, 2017, corporate net operating losses generated beginning in 2018 cannot be carried back but are carried forward indefinitely.
In the accompanying statements of operations research and development credits are included in other income, and taxes, other than income taxes, are included in general and administrative costs.
These net operating losses could offset only up to 80% of taxable income in future years (pre-2018 net operating losses could continue to offset taxable income with no limitation). The approximate $5.4 million, $4.5 million, and $3.1 million of Federal net operating loss generated in 2020, 2019, and 2018, respectively, is subject to this limitation.
The Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”), enacted and signed into law by President Trump on March 27, 2020 in response to the COVID-19 pandemic, temporarily suspends changes to the net operating loss rules made in the TCJA. The first change is that it temporarily removes the taxable income limitation, allowing net operating loss carryforwards to fully offset income. For tax years beginning after December 31, 2017 and before January 1, 2021, the Company is eligible to offset 100% of taxable income in years prior to January 1, 2021 and 80% of taxable income in years beginning January 1, 2021.
Since the ability to use net operating loss carryforwards and credits in the future is uncertain, they are recorded as deferred tax assets with a full valuation allowance. They will continue to be recognized in the years that they are utilized.
The components of the deferred tax assets at September 30, 2021 and 2020 are comprised of:
|
2021 |
2020 |
|||||||
|
Federal net operating carryforward |
$ |
25,035,200 |
|
$ |
18,552,900 |
|
||
|
Share-based compensation |
|
1,710,600 |
|
|
1,446,500 |
|
||
|
Anticipated marginal tax rate |
|
21 |
% |
|
21 |
% |
||
|
|
5,616,600 |
|
|
4,199,800 |
|
|||
|
Federal research and development credit |
|
200,500 |
|
|
200,500 |
|
||
|
Total Federal deferred tax assets |
|
5,817,100 |
|
|
4,400,300 |
|
||
|
Valuation allowance |
|
(5,817,100 |
) |
|
(4,400,300 |
) |
||
|
Net deferred tax asset |
$ |
— |
|
$ |
— |
|
||
|
2021 |
2020 |
|||||||
|
Connecticut net operating carryforward |
$ |
24,970,400 |
|
$ |
18,503,100 |
|
||
|
Anticipated marginal tax rate |
|
7.5 |
% |
|
7.5 |
% |
||
|
|
1,872,800 |
|
|
1,387,700 |
|
|||
|
State research and development credit |
|
204,200 |
|
|
136,000 |
|
||
|
Total Connecticut deferred tax assets |
|
2,077,000 |
|
|
1,523,700 |
|
||
|
Valuation allowance |
|
(2,077,000 |
) |
|
(1,523,700 |
) |
||
|
Net deferred tax asset |
$ |
— |
|
$ |
— |
|
||
F-42
INTENSITY THERAPEUTICS, INC.
Notes to the Financial Statements
September 30, 2021 and 2020
Note O — Retirement Plan — Defined Contribution
The Company maintains a defined contribution plan for all employees age 21 and older who have completed one month of service. This 401K plan began for payrolls after July 1, 2017. The Company makes a matching contribution equal to 100% of an employee’s contribution, up to 3% of an employee’s eligible earnings. The Company match is vested after one year of service. Retirement expense for this plan was approximately $34,000 and $24,000 for the nine months ended September 30, 2021 and 2020, respectively.
Note P — Subsequent Events
The Company evaluated subsequent events for financial reporting purposes through October 28, 2021, the date which the unaudited financial statements were issued to determine whether any events occurred that required adjustment to or disclosure in the accompanying financial statements. The Company concluded that no additional subsequent events required disclosure in these financial statements other than those disclosed in these notes to the financial statements.
F-43
Through and including , 2022 (the 25th day after the commencement of our initial public offering), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
Shares of Common Stock
Intensity Therapeutics, Inc.
____________________
PROSPECTUS
____________________
Sole Book-Running Manager
A.G.P.
Co-Manager
Brookline Capital Markets
a division of Arcadia Securities, LLC
, 2022
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth the estimated expenses payable by us in connection with the sale and distribution of the securities registered hereby, other than underwriting discounts or commissions. All amounts are estimates except for the SEC registration fee and the FINRA filing fee.
|
SEC registration fee |
$ |
1,759 |
|
|
FINRA filing fee |
|
3,950 |
|
|
Stock exchange listing fees |
|
75,000 |
|
|
Printing and engraving expenses |
|
40,000 |
|
|
Accounting fees and expenses |
|
155,000 |
|
|
Legal fees and expenses |
|
600,000 |
|
|
Transfer agent and registrar fees |
|
5,500 |
|
|
Miscellaneous fees and expenses |
|
18,791 |
|
|
TOTAL |
$ |
900,000 |
Item 14. Indemnification of Directors and Officers.
Section 145 of the General Corporation Law of the State of Delaware provides as follows:
A corporation shall have the power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interest of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. The termination of any action, suit or proceeding by judgment, order, settlement, conviction or upon a plea of nolo contendere or its equivalent shall not, of itself, create a presumption that the person did not act in good faith and in a manner which the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had reasonable cause to believe that his conduct was unlawful.
A corporation shall have the power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses (including attorneys’ fees) actually and reasonably incurred by him in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation and except that no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or the court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
As permitted by the Delaware General Corporation Law, we have included in our amended and restated certificate of incorporation a provision to eliminate the personal liability of our directors for monetary damages for breach of their fiduciary duties as directors, subject to certain exceptions. In addition, our amended and restated certificate of incorporation provides that we are required to indemnify our officers and directors under certain circumstances,
II-1
including those circumstances in which indemnification would otherwise be discretionary, and we are required to advance expenses to our officers and directors as incurred in connection with proceedings against them for which they may be indemnified.
We intend to enter into indemnification agreements with our directors and officers. These agreements will provide broader indemnity rights than those provided under the Delaware General Corporation Law and our amended and restated certificate of incorporation. The indemnification agreements are not intended to deny or otherwise limit third-party or derivative suits against us or our directors or officers, but to the extent a director or officer were entitled to indemnity or contribution under the indemnification agreement, the financial burden of a third-party suit would be borne by us, and we would not benefit from derivative recoveries against the director or officer. Such recoveries would accrue to our benefit but would be offset by our obligations to the director or officer under the indemnification agreement.
The underwriting agreement provides that the underwriters are obligated, under certain circumstances, to indemnify our directors, officers and controlling persons against certain liabilities, including liabilities under the Securities Act. Reference is made to the form of underwriting agreement filed as Exhibit 1.1 hereto.
We maintain directors’ and officers’ liability insurance for the benefit of our directors and officers.
Item 15. Recent Sales of Unregistered Securities.
Set forth below is information regarding unregistered securities issued by us within the past three years. Also included is the consideration received by us for such unregistered securities and information relating to the section of the Securities Act, or rule of the Securities and Exchange Commission, under which exemption from registration was claimed.
• Between May 2018 and September 2018, the Company sold 1,449,113 shares of Series B preferred stock in a private placement for an aggregate purchase price of $6,521,009.
• Between December 2019 and June 2020, the Company sold 1,800,606 shares of Series C preferred stock in a private placement for an aggregate purchase price of $10,353,485. In connection with such sales, the Company also issued warrants that may be exercised for up to 60,000 shares of Series C preferred stock.
• On February 1, 2020, in consideration for services related to sales of our Series C preferred stock, the Company sold 14,217 shares of common stock in a private placement to B Riley FBR for an aggregate purchase price of $81,748.
• On September 20, 2021, the Company entered into convertible debt agreement with a holder for aggregate principal of $2,000,000. The outstanding principal balance together with the unpaid and accrued interest of the notes will be automatically converted upon the completion of this offering at a conversion price equal to 75% of our initial public offering price.
• During 2018, 2019, 2020 and 2021, the Company issued 265,000, 280,000, 359,500 and 428,000 options, respectively, to employees pursuant to the 2103 Stock and Option Plan.
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits
|
Exhibit Number |
Description of Exhibit |
|
|
1.1+ |
||
|
3.1+ |
Certificate of Incorporation of the Registrant, as currently in effect |
|
|
3.2+ |
||
|
3.3+ |
||
|
3.4+ |
||
|
4.1+ |
Specimen Common Stock Certificate evidencing the shares of Common Stock |
|
|
5.1+ |
||
|
10.1+ |
II-2
|
Exhibit Number |
Description of Exhibit |
|
|
10.2+# |
||
|
10.3+# |
||
|
10.4+# |
Amended and Restated Employment Agreement between the Registrant and Lewis H. Bender |
|
|
10.5+# |
Employment Agreement, dated June 21, 2019, between the Registrant and Rebecca “Peggi” Drain |
|
|
10.6+# |
Employment Agreement, dated August 25, 2014, between the Registrant and Ian B. Walters |
|
|
10.7+† |
||
|
10.8+† |
||
|
10.9+† |
||
|
21.1+ |
||
|
23.1 |
||
|
23.2+ |
Consent of McDermott Will & Emery LLP (included in Exhibit 5.1) |
|
|
24.1+ |
____________
+ Previously filed
† Certain information has been excluded from the exhibit because it both (i) is not material and (ii) would likely cause competitive harm to the Registrant if publicly disclosed.
# Indicates a management contract or any compensatory plan, contract or arrangement.
(b) Financial Statement Schedules
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
Item 17. Undertakings.
The undersigned Registrant hereby undertakes:
(1) That for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933 shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) That for the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) For the purpose of determining liability under the Securities Act of 1933 to any purchaser, if the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
II-3
(4) The undersigned Registrant undertakes that in a primary offering of securities of the undersigned Registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned Registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned Registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned Registrant or used or referred to by the undersigned Registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned Registrant or its securities provided by or on behalf of the undersigned Registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned Registrant to the purchaser.
(5) To provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
(6) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Westport, CT on the 7th day of January, 2022.
|
Intensity Therapeutics, Inc. |
||||||
|
By: |
/s/ Lewis H. Bender |
|||||
|
Name: |
Lewis H. Bender |
|||||
|
Title: |
President and Chief Executive Officer, Chairman |
|||||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the 7th day of January, 2022.
|
Signature |
Title |
|
|
/s/ Lewis H. Bender |
President and Chief Executive Officer, Chairman |
|
|
Lewis H. Bender |
(Principal Executive Officer) |
|
|
/s/ James M. Ahlers |
Chief Financial Officer |
|
|
James M. Ahlers |
(Principal Financial Officer) |
|
|
/s/ John Wesolowski |
Principal Accounting Officer and Controller |
|
|
John Wesolowski |
(Principal Accounting Officer) |
|
|
* |
Director |
|
|
Dr. Declan Doogan |
||
|
* |
Director |
|
|
Dr. Emer Leahy |
||
|
* |
Director |
|
|
Dr. Mark A. Goldberg |
|
*By: |
/s/ Lewis H. Bender |
|||
|
Lewis H. Bender |
||||
|
Attorney-in-Fact |
II-5